Yu Lili, Chen Yajun, Xing Ligang
School of Medicine and Life Sciences, Shandong Academy of Medical Sciences, University of Jinan; Department of Radiation Oncology, Shandong Cancer Hospital Affiliated to Shandong University, Jinan, People's Republic of China.
Department of Radiation Oncology, Shandong Cancer Hospital Affiliated to Shandong University, Jinan, People's Republic of China.
Onco Targets Ther. 2017 Mar 20;10:1687-1694. doi: 10.2147/OTT.S121521. eCollection 2017.
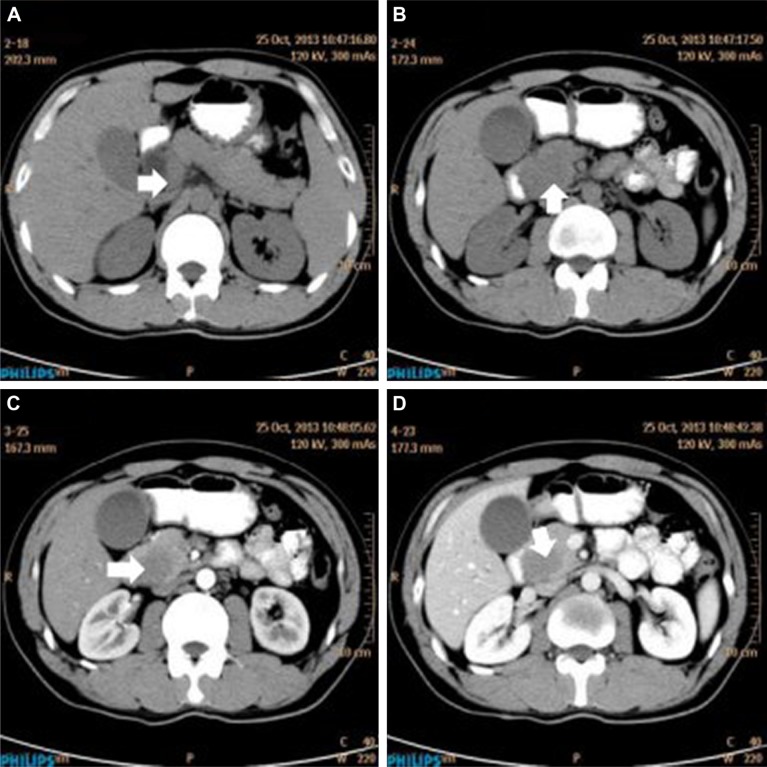
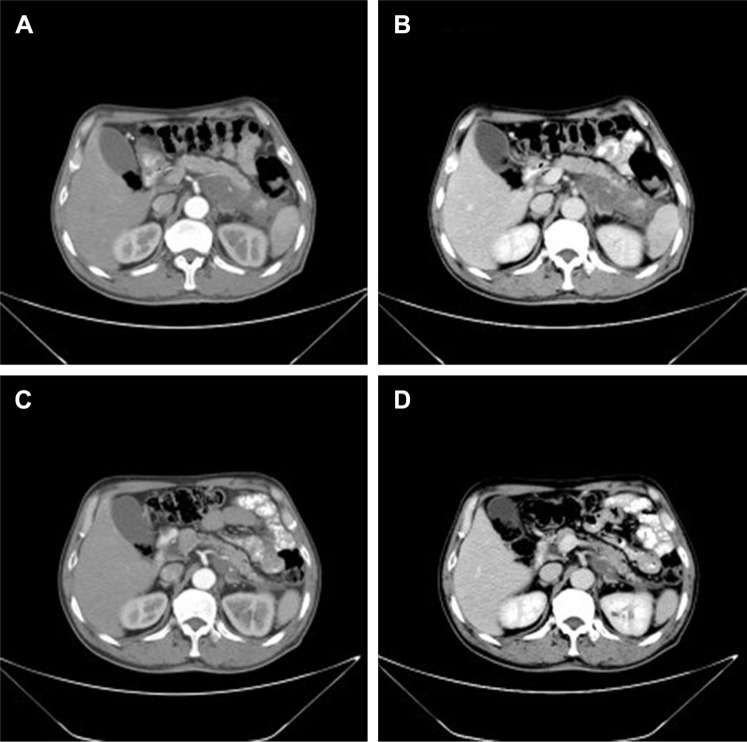
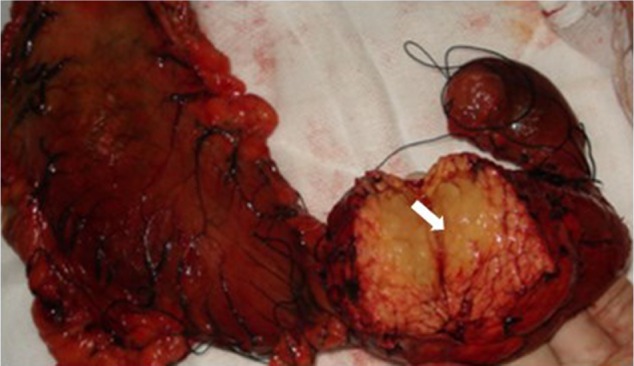
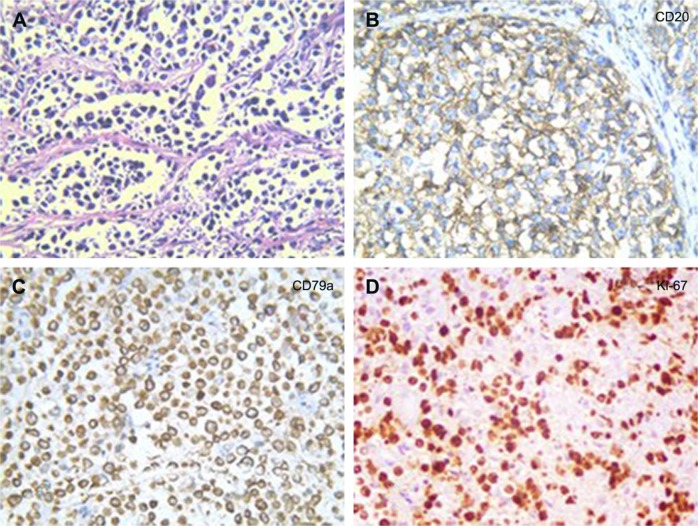
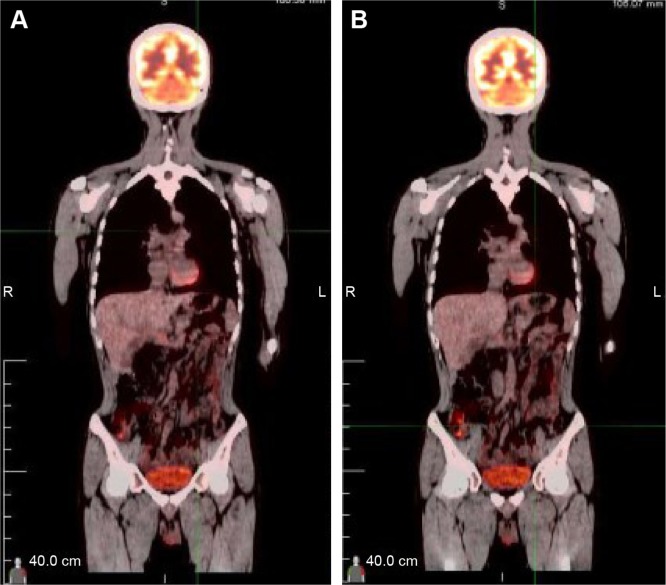
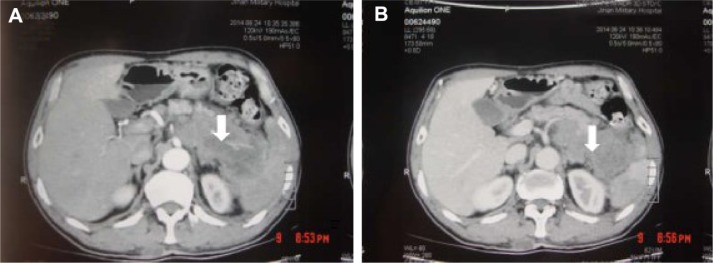
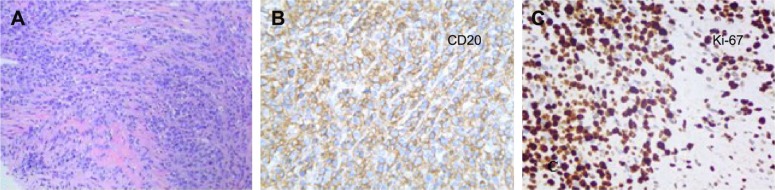
Primary pancreatic lymphoma (PPL) is an extremely rare disease, with only a few cases reported in the literature. Clinical manifestations of PPL are often nonspecific and may mimic other pancreatic diseases. Because of the limited experience of PPL, clinicopathological features, differential diagnosis, optimal therapy, and outcomes are not well defined. We described two cases diagnosed as PPL and confirmed by histological examination and immunohistochemical analysis. Case 1 was a young man with obstructive jaundice and upper abdominal malaise mimicking a pancreatic adenocarcinoma. A computed tomography (CT) scan revealed a diffuse heterogeneous mass in the head of the pancreas along with dilated bile ducts, no dilated pancreatic duct, no liver or splenic involvement, or evident retroperitoneal adenopathies. The patient underwent a pancreatico-duodenectomy, and the postoperative histopathology confirmed diffuse large B-cell non-Hodgkin lymphoma. Postoperatively, he received six courses of the CHOP regimen (cyclophosphamide, doxorubicin, vincristine, and prednisolone). Case 2 was an older man with left flank pain. A CT confirmed a mass with irregular margins at the tail of the pancreas and the hilum of the spleen. The mass was heterogeneous, with no clear boundary between lesions, spleen, stomach, and pancreas, with nearby blood vessels wrapped around it, and multiple enlarged lymph nodes in the abdominal cavity. A CT-guided biopsy was performed. The immunohistological findings of the specimen revealed a diffuse large B-cell lymphoma. The size of the tumor was significantly reduced after four cycles of the CHOP chemotherapy regimen. These two cases were different in clinical manifestation, location, and treatment. We reviewed the literature and discussed the clinicopathological features, differential diagnosis, optimal therapy, and outcomes of this neoplasm.
原发性胰腺淋巴瘤(PPL)是一种极其罕见的疾病,文献中仅报道了少数病例。PPL的临床表现通常不具有特异性,可能与其他胰腺疾病相似。由于PPL的经验有限,其临床病理特征、鉴别诊断、最佳治疗方法及预后尚不明确。我们描述了两例经组织学检查和免疫组化分析确诊为PPL的病例。病例1是一名年轻男性,表现为梗阻性黄疸和上腹部不适,类似胰腺腺癌。计算机断层扫描(CT)显示胰头有弥漫性不均匀肿块,伴有胆管扩张,胰管未扩张,无肝脏或脾脏受累,也无明显的腹膜后淋巴结肿大。患者接受了胰十二指肠切除术,术后组织病理学证实为弥漫性大B细胞非霍奇金淋巴瘤。术后,他接受了六个疗程的CHOP方案(环磷酰胺、阿霉素、长春新碱和泼尼松龙)治疗。病例2是一名老年男性,有左侧腰痛。CT证实胰腺尾部和脾门处有一个边缘不规则的肿块。肿块不均匀,病变与脾脏、胃和胰腺之间无明显边界,附近血管被其包裹,腹腔内有多个肿大的淋巴结。进行了CT引导下活检。标本的免疫组化结果显示为弥漫性大B细胞淋巴瘤。CHOP化疗方案四个周期后肿瘤大小明显缩小。这两例病例在临床表现、部位和治疗方面存在差异。我们回顾了文献并讨论了该肿瘤的临床病理特征(clinicopathological features)、鉴别诊断、最佳治疗方法及预后。