Baburajeev C P, Mohan Chakrabhavi Dhananjaya, Rangappa Shobith, Mason Daniel J, Fuchs Julian E, Bender Andreas, Barash Uri, Vlodavsky Israel, Rangappa Kanchugarakoppal S
Laboratory of Chemical Biology, Department of Chemistry, Bangalore University, Central College Campus, Palace Road, Bangalore, 560001, India.
Department of Studies in Chemistry, University of Mysore, Manasagangotri, Mysore, 570006, India.
BMC Cancer. 2017 Mar 31;17(1):235. doi: 10.1186/s12885-017-3214-8.
Expression and activity of heparanase, an endoglycosidase that cleaves heparan sulfate (HS) side chains of proteoglycans, is associated with progression and poor prognosis of many cancers which makes it an attractive drug target in cancer therapeutics.
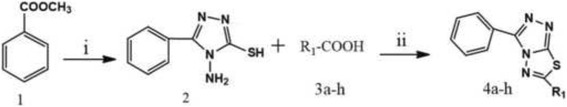
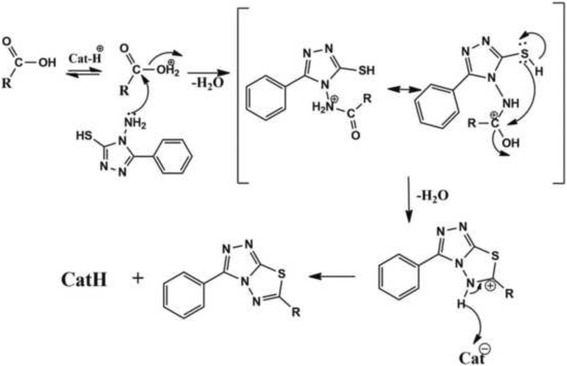
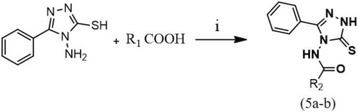
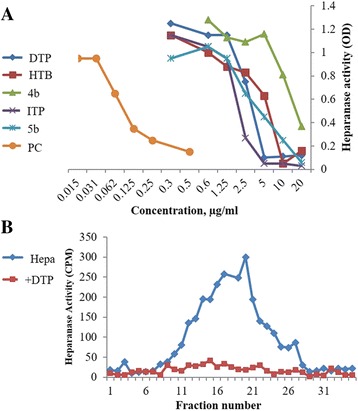
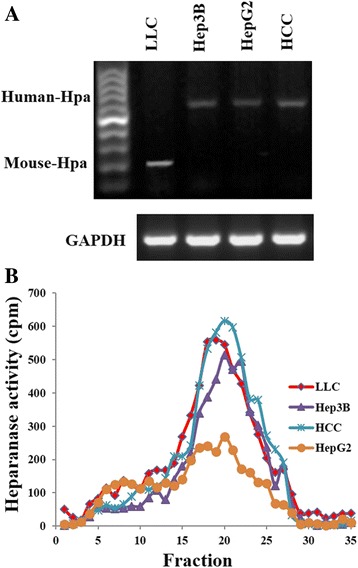
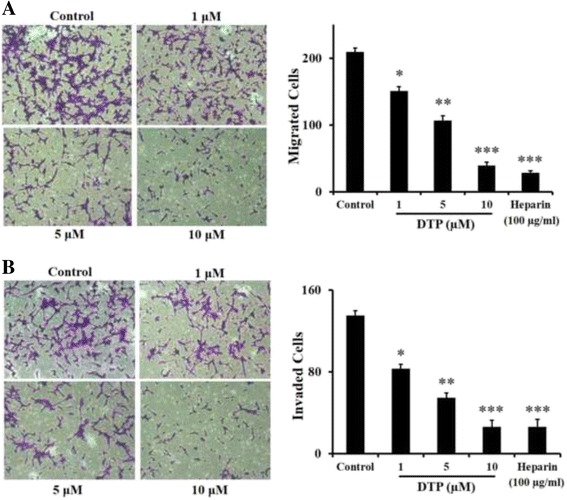
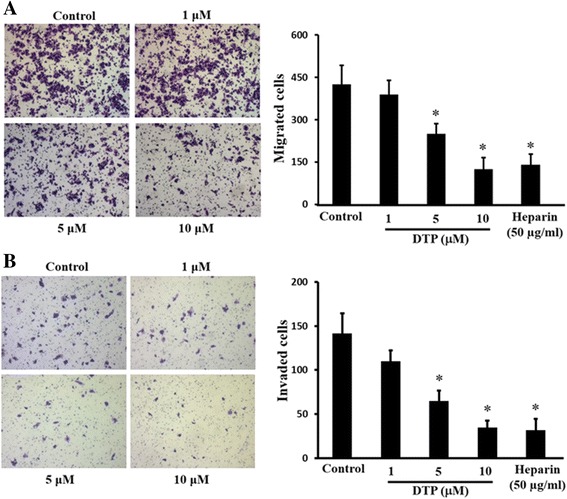
In the present work, we report the in vitro screening of a library of 150 small molecules with the scaffold bearing quinolones, oxazines, benzoxazines, isoxazoli(di)nes, pyrimidinones, quinolines, benzoxazines, and 4-thiazolidinones, thiadiazolo[3,2-a]pyrimidin-5-one, 1,2,4-triazolo-1,3,4-thiadiazoles, and azaspiranes against the enzymatic activity of human heparanase. The identified lead compounds were evaluated for their heparanase-inhibiting activity using sulfate [S] labeled extracellular matrix (ECM) deposited by cultured endothelial cells. Further, anti-invasive efficacy of lead compound was evaluated against hepatocellular carcinoma (HepG2) and Lewis lung carcinoma (LLC) cells.
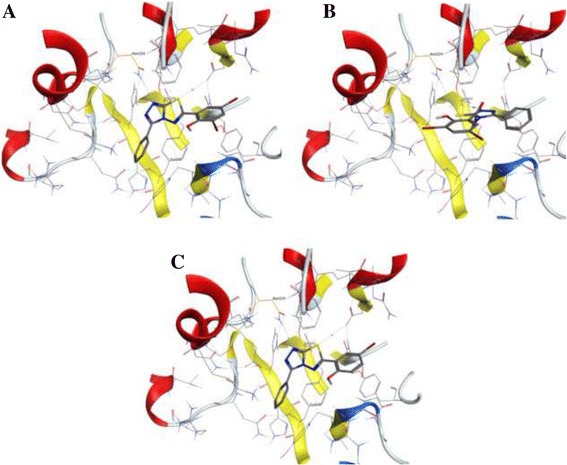
Among the 150 compounds screened, we identified 1,2,4-triazolo-1,3,4-thiadiazoles bearing compounds to possess human heparanase inhibitory activity. Further analysis revealed 2,4-Diiodo-6-(3-phenyl-[1, 2, 4]triazolo[3,4-b][1, 3, 4]thiadiazol-6yl)phenol (DTP) as the most potent inhibitor of heparanase enzymatic activity among the tested compounds. The inhibitory efficacy was demonstrated by a colorimetric assay and further validated by measuring the release of radioactive heparan sulfate degradation fragments from [S] labeled extracellular matrix. Additionally, lead compound significantly suppressed migration and invasion of LLC and HepG2 cells with IC value of ~5 μM. Furthermore, molecular docking analysis revealed a favourable interaction of triazolo-thiadiazole backbone with Asn-224 and Asp-62 of the enzyme.
Overall, we identified biologically active heparanase inhibitor which could serve as a lead structure in developing compounds that target heparanase in cancer.
硫酸乙酰肝素酶是一种可切割蛋白聚糖硫酸乙酰肝素(HS)侧链的内切糖苷酶,其表达和活性与多种癌症的进展及不良预后相关,这使其成为癌症治疗中一个有吸引力的药物靶点。
在本研究中,我们报告了对一个包含150种小分子的文库进行体外筛选的结果,这些小分子的骨架含有喹诺酮、恶嗪、苯并恶嗪、异恶唑(二)酮、嘧啶酮、喹啉、苯并恶嗪、4-噻唑烷酮、噻二唑并[3,2-a]嘧啶-5-酮、1,2,4-三唑并-1,3,4-噻二唑以及氮杂螺环化合物,以检测它们对人硫酸乙酰肝素酶酶活性的影响。使用由培养的内皮细胞沉积的硫酸盐[S]标记的细胞外基质(ECM),对鉴定出的先导化合物的硫酸乙酰肝素酶抑制活性进行评估。此外,评估了先导化合物对肝癌细胞(HepG2)和Lewis肺癌细胞(LLC)的抗侵袭效果。
在筛选的150种化合物中,我们鉴定出含1,2,4-三唑并-1,3,4-噻二唑的化合物具有人硫酸乙酰肝素酶抑制活性。进一步分析显示,2,4-二碘-6-(3-苯基-[1,2,4]三唑并[3,4-b][1,3,4]噻二唑-6-基)苯酚(DTP)是测试化合物中硫酸乙酰肝素酶酶活性最有效的抑制剂。通过比色法测定证明了其抑制效果,并通过测量从[S]标记的细胞外基质中释放的放射性硫酸乙酰肝素降解片段进一步验证。此外,先导化合物显著抑制LLC和HepG2细胞的迁移和侵袭,IC值约为5 μM。此外,分子对接分析显示三唑并噻二唑骨架与该酶的Asn-224和Asp-62有良好的相互作用。
总体而言,我们鉴定出了具有生物活性的硫酸乙酰肝素酶抑制剂,其可作为开发针对癌症中硫酸乙酰肝素酶的化合物的先导结构。