Martín-Durán José M, Ryan Joseph F, Vellutini Bruno C, Pang Kevin, Hejnol Andreas
Sars International Centre for Marine Molecular Biology, University of Bergen, Bergen 5006, Norway.
Whitney Laboratory for Marine Bioscience, University of Florida, St. Augustine, Florida 32080, USA.
Genome Res. 2017 Jul;27(7):1263-1272. doi: 10.1101/gr.216226.116. Epub 2017 Apr 11.
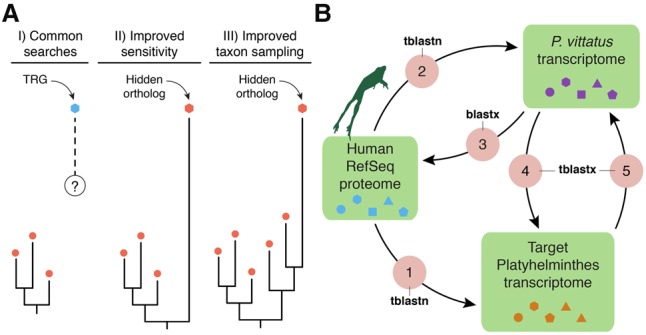
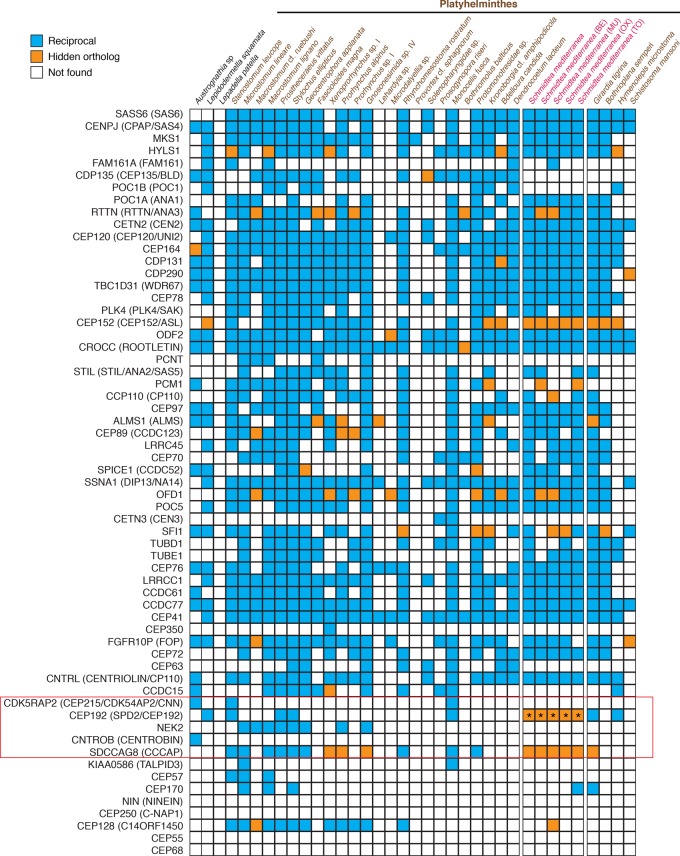
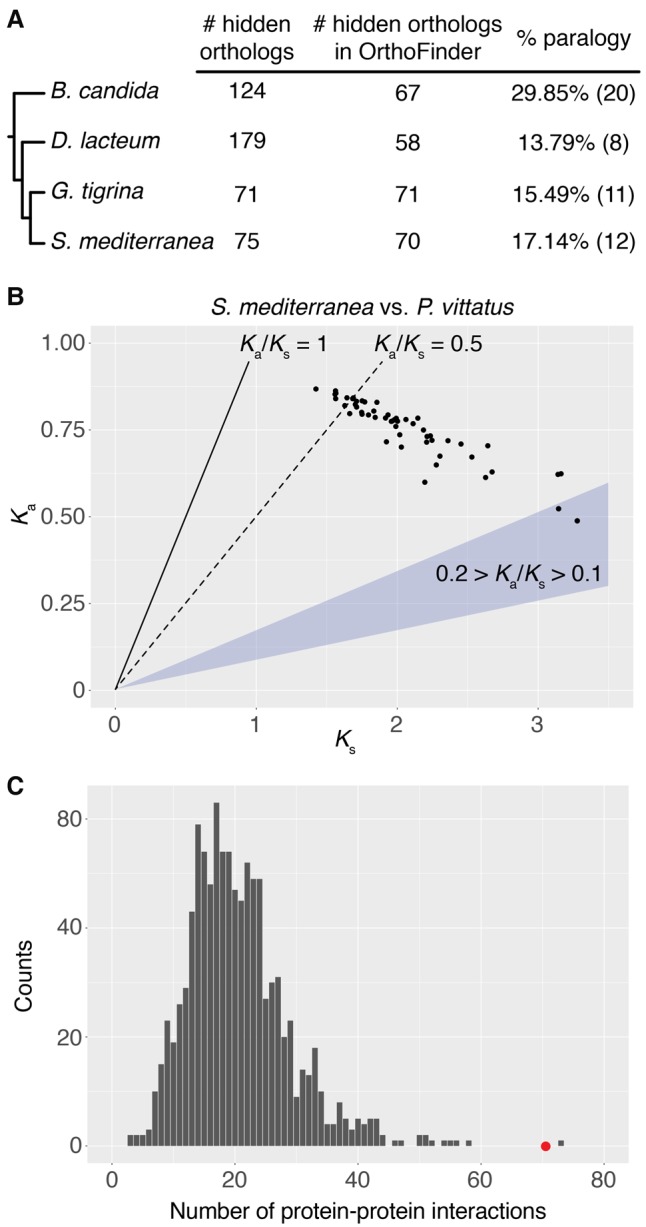
Gains and losses shape the gene complement of animal lineages and are a fundamental aspect of genomic evolution. Acquiring a comprehensive view of the evolution of gene repertoires is limited by the intrinsic limitations of common sequence similarity searches and available databases. Thus, a subset of the gene complement of an organism consists of hidden orthologs, i.e., those with no apparent homology to sequenced animal lineages-mistakenly considered new genes-but actually representing rapidly evolving orthologs or undetected paralogs. Here, we describe Leapfrog, a simple automated BLAST pipeline that leverages increased taxon sampling to overcome long evolutionary distances and identify putative hidden orthologs in large transcriptomic databases by transitive homology. As a case study, we used 35 transcriptomes of 29 flatworm lineages to recover 3427 putative hidden orthologs, some unidentified by OrthoFinder and HaMStR, two common orthogroup inference algorithms. Unexpectedly, we do not observe a correlation between the number of putative hidden orthologs in a lineage and its "average" evolutionary rate. Hidden orthologs do not show unusual sequence composition biases that might account for systematic errors in sequence similarity searches. Instead, gene duplication with divergence of one paralog and weak positive selection appear to underlie hidden orthology in Platyhelminthes. By using Leapfrog, we identify key centrosome-related genes and homeodomain classes previously reported as absent in free-living flatworms, e.g., planarians. Altogether, our findings demonstrate that hidden orthologs comprise a significant proportion of the gene repertoire in flatworms, qualifying the impact of gene losses and gains in gene complement evolution.
基因的获得与丢失塑造了动物谱系的基因组成,是基因组进化的一个基本方面。由于常见序列相似性搜索和现有数据库的内在局限性,全面了解基因库的进化受到限制。因此,生物体基因组成的一个子集由隐藏的直系同源基因组成,即那些与已测序动物谱系没有明显同源性的基因——被错误地认为是新基因——但实际上代表快速进化的直系同源基因或未检测到的旁系同源基因。在这里,我们描述了Leapfrog,这是一种简单的自动化BLAST流程,它利用增加的分类群采样来克服长进化距离,并通过传递同源性在大型转录组数据库中识别推定的隐藏直系同源基因。作为一个案例研究,我们使用了29个扁虫谱系的35个转录组来恢复3427个推定的隐藏直系同源基因,其中一些未被两种常见的直系同源组推断算法OrthoFinder和HaMStR识别。出乎意料的是,我们没有观察到一个谱系中推定的隐藏直系同源基因数量与其“平均”进化速率之间的相关性。隐藏的直系同源基因没有显示出可能解释序列相似性搜索中系统误差的异常序列组成偏差。相反,一个旁系同源基因发生分歧的基因复制和弱正选择似乎是扁形动物隐藏直系同源性的基础。通过使用Leapfrog,我们鉴定出了之前报道在自由生活扁虫(如涡虫)中不存在的关键中心体相关基因和同源异型域类别。总之,我们的研究结果表明,隐藏的直系同源基因在扁虫的基因库中占很大比例,这限定了基因获得和丢失对基因组成进化的影响。