Aydınok Yeşim, Oymak Yeşim, Atabay Berna, Aydoğan Gönül, Yeşilipek Akif, Ünal Selma, Kılınç Yurdanur, Oflaz Banu, Akın Mehmet, Vergin Canan, Sezgin Evim Melike, Çalışkan Ümran, Ünal Şule, Bay Ali, Kazancı Elif, İleri Talia, Atay Didem, Patıroğlu Türkan, Kahraman Selda, Söker Murat, Akcan Mediha, Akdeniz Aydan, Büyükavcı Mustafa, Alanoğlu Güçhan, Bör Özcan, Soyer Nur, Özdemir Karadaş Nihal, Uysalol Ezgi, Türker Meral, Akçay Arzu, Ocak Süheyla, Güneş Adalet Meral, Tokgöz Hüseyin, Ünal Elif, Tiftik Naci, Karakaş Zeynep
Hemoglobinopathy Study Group, Turkey.
Turk J Haematol. 2018 Mar 1;35(1):12-18. doi: 10.4274/tjh.2017.0039. Epub 2017 Apr 13.
The Turkish Society of Pediatric Hematology set up a National Hemoglobinopathy Registry to demonstrate the demographic and disease characteristics of patients and assess the efficacy of a hemoglobinopathy control program (HCP) over 10 years in Turkey.
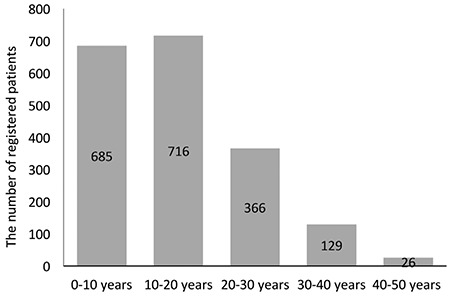
A total of 2046 patients from 27 thalassemia centers were registered, of which 1988 were eligible for analysis. This cohort mainly comprised patients with β-thalassemia major (n=1658, 83.4%) and intermedia (n=215, 10.8%).
The majority of patients were from the coastal areas of Turkey. The high number of patients in Southeastern Anatolia was due to that area having the highest rates of consanguineous marriage and fertility. The most common 11 mutations represented 90% of all β-thalassemia alleles and 47% of those were IVS1-110(G->A) mutations. The probability of undergoing splenectomy within the first 10 years of life was 20%, a rate unchanged since the 1980s. Iron chelators were administered as monotherapy regimens in 95% of patients and deferasirox was prescribed in 81.3% of those cases. Deferasirox administration was the highest (93.6%) in patients aged <10 years. Of the thalassemia major patients, 5.8% had match-related hemopoietic stem cell transplantation with a success rate of 77%. Cardiac disease was detected as a major cause of death and did not show a decreasing trend in 5-year cohorts since 1999.
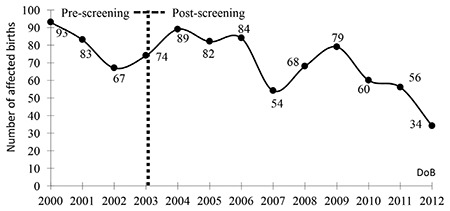
While the HCP has been implemented since 2003, the affected births have shown a consistent decrease only after 2009, being at lowest 34 cases per year. This program failure resulted from a lack of premarital screening in the majority of cases. Additional problems were unawareness of the risk and misinformation of the at-risk couples. In addition, prenatal diagnosis was either not offered to or was not accepted by the at-risk families. This study indicated that a continuous effort is needed for optimizing the management of thalassemia and the development of strategies is essential for further achievements in the HCP in Turkey.
土耳其儿科学血液学学会设立了一个全国血红蛋白病登记处,以展示患者的人口统计学和疾病特征,并评估土耳其一项血红蛋白病控制项目(HCP)在10年期间的成效。
来自27个地中海贫血中心的2046例患者进行了登记,其中1988例符合分析条件。该队列主要包括重型β地中海贫血患者(n = 1658,83.4%)和中间型β地中海贫血患者(n = 215,10.8%)。
大多数患者来自土耳其沿海地区。安纳托利亚东南部患者数量较多是因为该地区近亲结婚和生育率最高。最常见的11种突变占所有β地中海贫血等位基因的90%,其中47%为IVS1-110(G→A)突变。在生命的前10年内接受脾切除术的概率为20%,自20世纪80年代以来这一比例未变。95%的患者采用铁螯合剂单一疗法,其中81.3%的患者使用地拉罗司。年龄<10岁的患者中地拉罗司的使用率最高(93.6%)。在重型地中海贫血患者中,5.8%进行了匹配相关造血干细胞移植,成功率为77%。心脏病被检测为主要死亡原因,自1999年以来在5年队列中未呈下降趋势。
虽然自2003年起实施了HCP,但受影响的出生数仅在2009年后持续下降,最低为每年34例。该项目失败的原因是大多数情况下缺乏婚前筛查。其他问题包括对风险的认识不足以及对高危夫妇的错误信息。此外,高危家庭要么未获得产前诊断,要么不接受产前诊断服务。这项研究表明,需要持续努力优化地中海贫血的管理,制定策略对于土耳其HCP取得进一步成果至关重要。