Pavlidis Pavlos, Alachiotis Nikolaos
Institute of Computer Science, Foundation for Research and Technology-Hellas, 70013 Crete, Greece.
J Biol Res (Thessalon). 2017 Apr 8;24:7. doi: 10.1186/s40709-017-0064-0. eCollection 2017 Dec.
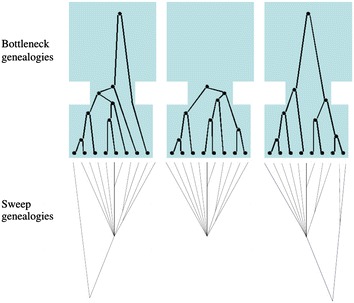
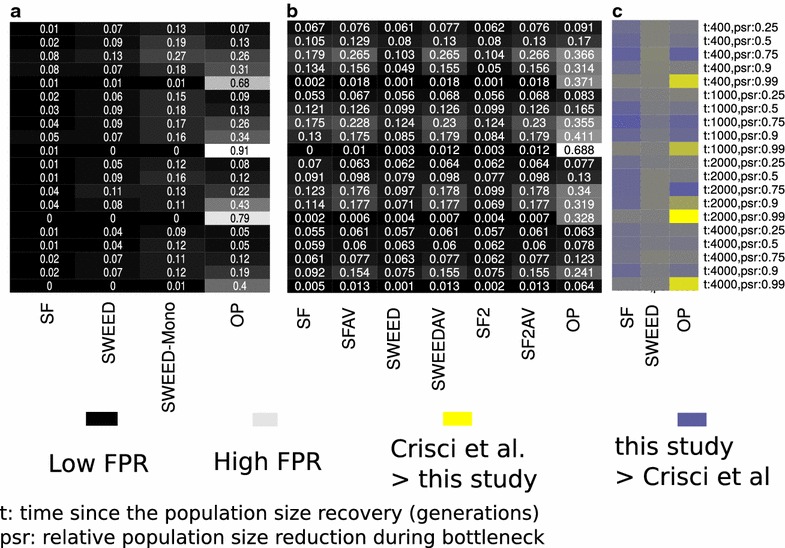
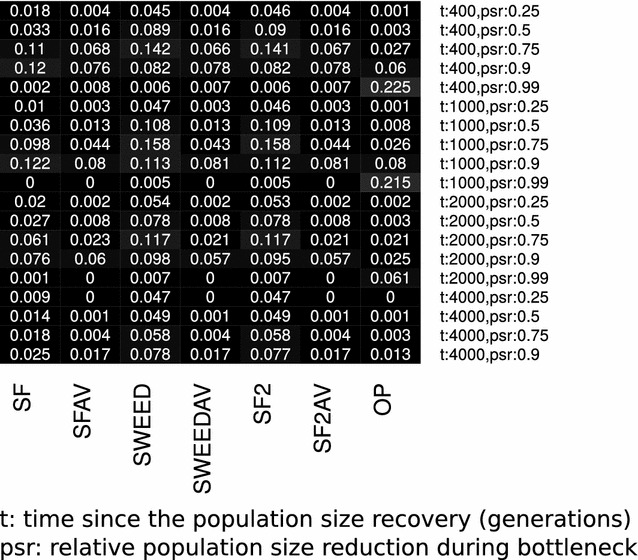
Positive selection occurs when an allele is favored by natural selection. The frequency of the favored allele increases in the population and due to genetic hitchhiking the neighboring linked variation diminishes, creating so-called selective sweeps. Detecting traces of positive selection in genomes is achieved by searching for signatures introduced by selective sweeps, such as regions of reduced variation, a specific shift of the site frequency spectrum, and particular LD patterns in the region. A variety of methods and tools can be used for detecting sweeps, ranging from simple implementations that compute summary statistics such as Tajima's D, to more advanced statistical approaches that use combinations of statistics, maximum likelihood, machine learning etc. In this survey, we present and discuss summary statistics and software tools, and classify them based on the selective sweep signature they detect, i.e., SFS-based vs. LD-based, as well as their capacity to analyze whole genomes or just subgenomic regions. Additionally, we summarize the results of comparisons among four open-source software releases (SweeD, SweepFinder, SweepFinder2, and OmegaPlus) regarding sensitivity, specificity, and execution times. In equilibrium neutral models or mild bottlenecks, both SFS- and LD-based methods are able to detect selective sweeps accurately. Methods and tools that rely on LD exhibit higher true positive rates than SFS-based ones under the model of a single sweep or recurrent hitchhiking. However, their false positive rate is elevated when a misspecified demographic model is used to represent the null hypothesis. When the correct (or similar to the correct) demographic model is used instead, the false positive rates are considerably reduced. The accuracy of detecting the true target of selection is decreased in bottleneck scenarios. In terms of execution time, LD-based methods are typically faster than SFS-based methods, due to the nature of required arithmetic.
当一个等位基因受到自然选择的青睐时,就会发生正向选择。受青睐的等位基因在种群中的频率会增加,并且由于遗传搭便车效应,相邻的连锁变异会减少,从而产生所谓的选择性清除。通过搜索选择性清除引入的特征,如变异减少的区域、位点频率谱的特定偏移以及该区域特定的连锁不平衡模式,来检测基因组中的正向选择痕迹。可以使用多种方法和工具来检测选择性清除,从计算诸如 Tajima's D 等汇总统计量的简单实现,到使用统计量组合、最大似然法、机器学习等的更先进统计方法。在本次综述中,我们展示并讨论汇总统计量和软件工具,并根据它们检测到的选择性清除特征对其进行分类,即基于位点频率谱(SFS)的方法与基于连锁不平衡(LD)的方法,以及它们分析全基因组或仅亚基因组区域的能力。此外,我们总结了四个开源软件版本(SweeD、SweepFinder、SweepFinder2 和 OmegaPlus)在灵敏度、特异性和执行时间方面的比较结果。在平衡中性模型或轻度瓶颈情况下,基于 SFS 和基于 LD 的方法都能够准确检测选择性清除。在单次清除或反复搭便车模型下,依赖 LD 的方法比基于 SFS 的方法具有更高的真阳性率。然而,当使用错误指定的群体模型来表示零假设时,它们的假阳性率会升高。当使用正确(或与正确相似)的群体模型时,假阳性率会大幅降低。在瓶颈情况下,检测选择真实目标的准确性会降低。在执行时间方面,由于所需算法的性质,基于 LD 的方法通常比基于 SFS 的方法更快。