Wako Hiroshi, Abe Haruo
School of Social Sciences, Waseda University, Shinjuku, Tokyo 169-8050, Japan.
Department of Electrical Engineering, Nishinippon Institute of Technology, Miyako, Fukuoka 800-0394, Japan.
Biophys Physicobiol. 2016 Nov 18;13:263-279. doi: 10.2142/biophysico.13.0_263. eCollection 2016.
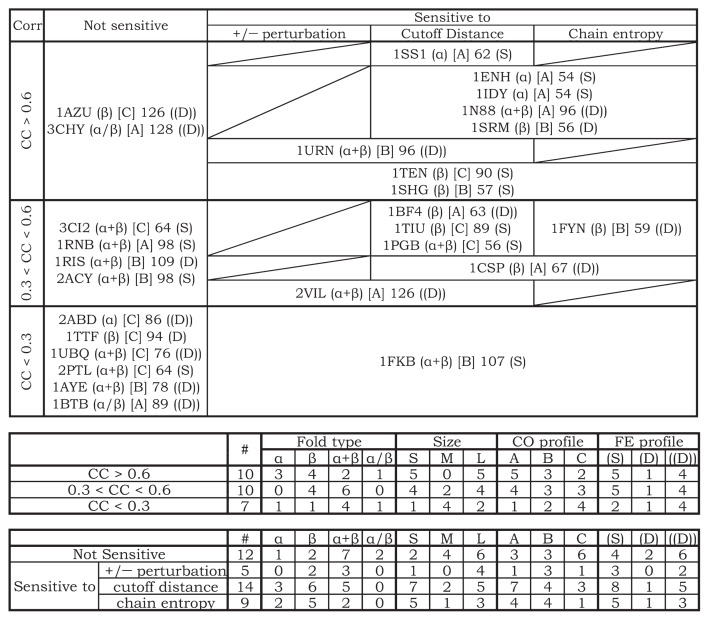
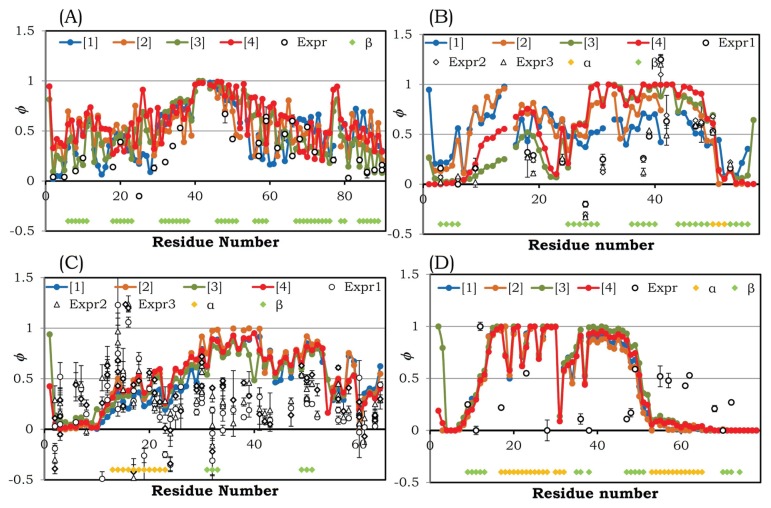
The Φ-value analysis approach provides information about transition-state structures along the folding pathway of a protein by measuring the effects of an amino acid mutation on folding kinetics. Here we compared the theoretically calculated Φ values of 27 proteins with their experimentally observed Φ values; the theoretical values were calculated using a simple statistical-mechanical model of protein folding. The theoretically calculated Φ values reflected the corresponding experimentally observed Φ values with reasonable accuracy for many of the proteins, but not for all. The correlation between the theoretically calculated and experimentally observed Φ values strongly depends on whether the protein-folding mechanism assumed in the model holds true in real proteins. In other words, the correlation coefficient can be expected to illuminate the folding mechanisms of proteins, providing the answer to the question of which model more accurately describes protein folding: the framework model or the nucleation-condensation model. In addition, we tried to characterize protein folding with respect to various properties of each protein apart from the size and fold class, such as the free-energy profile, contact-order profile, and sensitivity to the parameters used in the Φ-value calculation. The results showed that any one of these properties alone was not enough to explain protein folding, although each one played a significant role in it. We have confirmed the importance of characterizing protein folding from various perspectives. Our findings have also highlighted that protein folding is highly variable and unique across different proteins, and this should be considered while pursuing a unified theory of protein folding.
Φ值分析方法通过测量氨基酸突变对折叠动力学的影响,提供了蛋白质折叠途径中过渡态结构的信息。在此,我们将27种蛋白质的理论计算Φ值与其实验观测的Φ值进行了比较;理论值是使用蛋白质折叠的简单统计力学模型计算得出的。对于许多蛋白质,理论计算的Φ值能以合理的准确度反映相应的实验观测Φ值,但并非所有蛋白质都如此。理论计算值与实验观测值之间的相关性很大程度上取决于模型中假设的蛋白质折叠机制在实际蛋白质中是否成立。换句话说,相关系数有望阐明蛋白质的折叠机制,从而回答哪种模型能更准确描述蛋白质折叠的问题:框架模型还是成核凝聚模型。此外,我们试图从每种蛋白质除大小和折叠类别之外的各种特性方面来描述蛋白质折叠,比如自由能分布、接触序分布以及对Φ值计算中所用参数的敏感性。结果表明,尽管这些特性中的任何一个在蛋白质折叠中都起着重要作用,但仅靠其中任何一个特性都不足以解释蛋白质折叠。我们证实了从多个角度描述蛋白质折叠的重要性。我们的研究结果还突出表明,不同蛋白质的蛋白质折叠具有高度的变异性和独特性,在寻求蛋白质折叠统一理论时应考虑到这一点。