Kathiriya Jaymin J, Nakra Niyati, Nixon Jenna, Patel Puja S, Vaghasiya Vijay, Alhassani Ahmed, Tian Zhi, Allen-Gipson Diane, Davé Vrushank
Department of Pathology and Cell Biology, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA.
University of Miami, Coral Gables, FL 33124, USA.
Cell Death Discov. 2017 Apr 10;3:17010. doi: 10.1038/cddiscovery.2017.10. eCollection 2017.
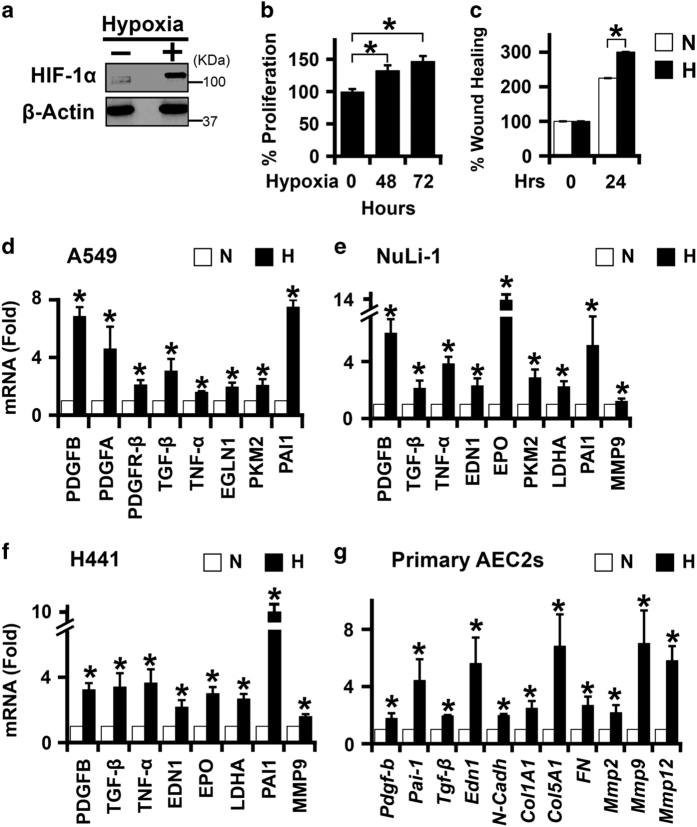
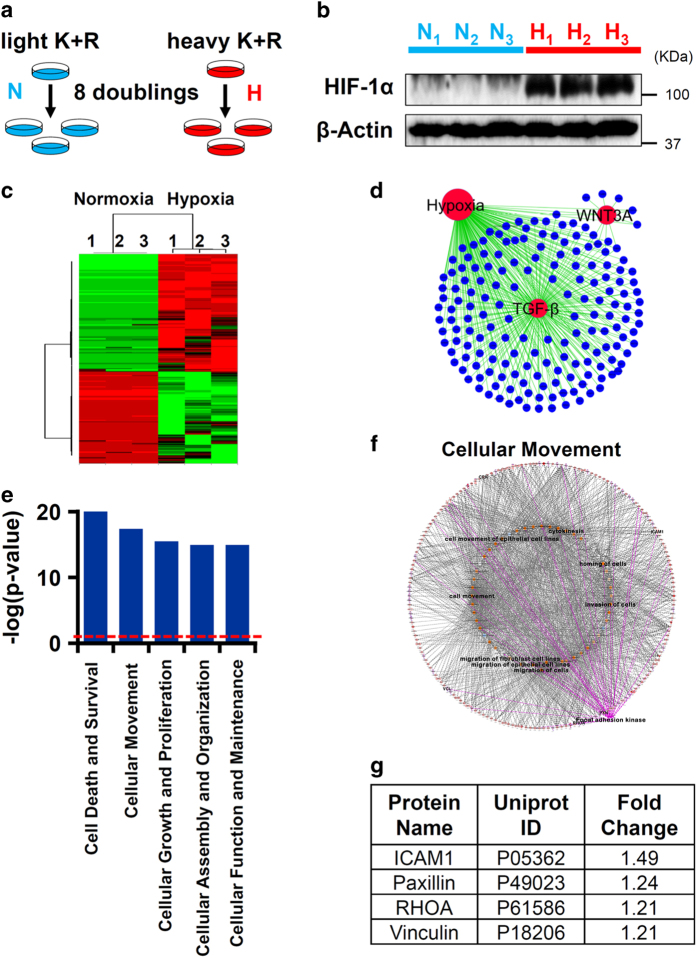
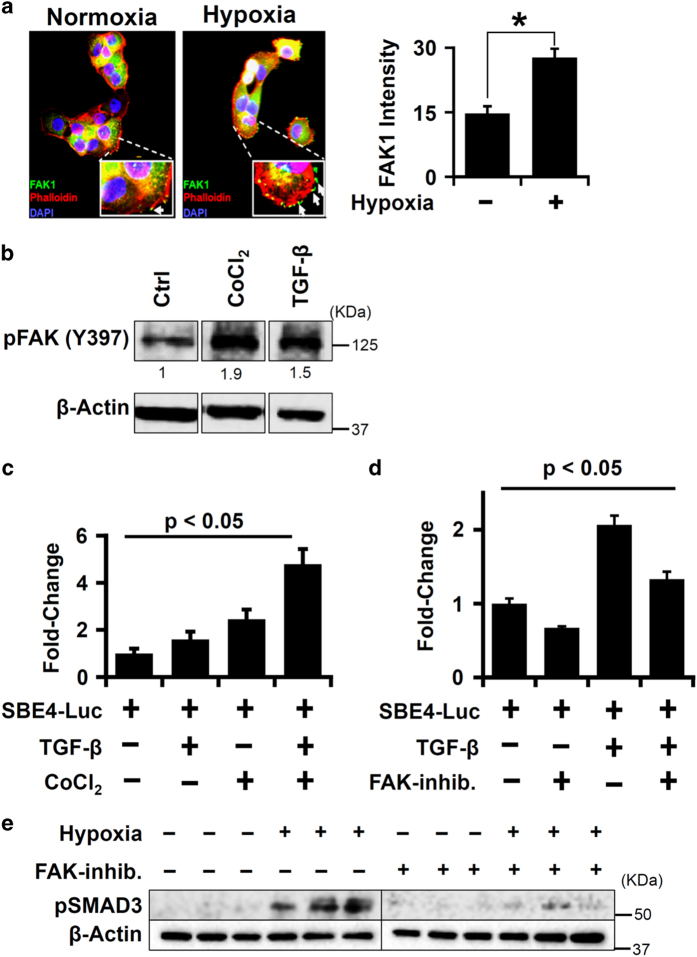
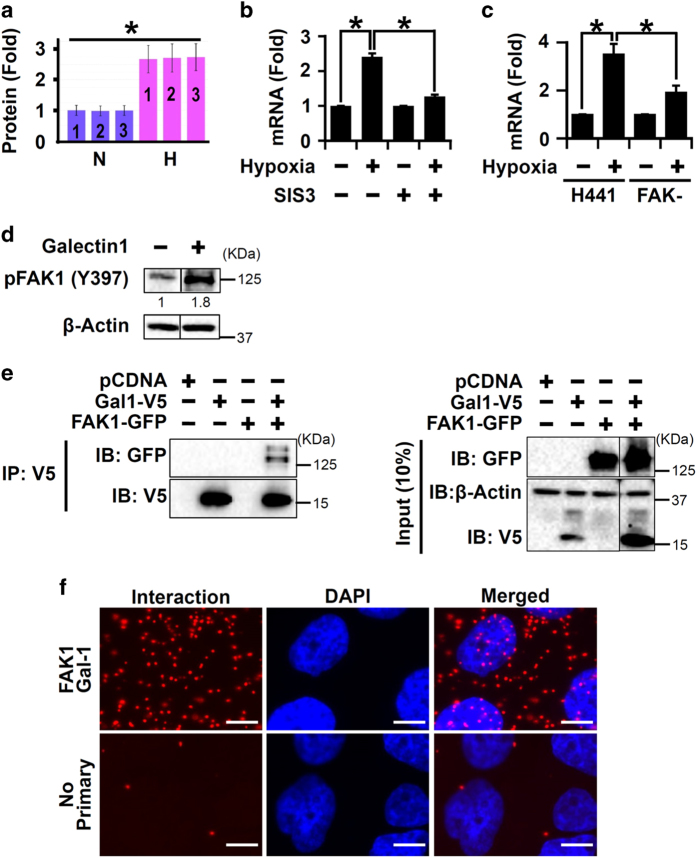
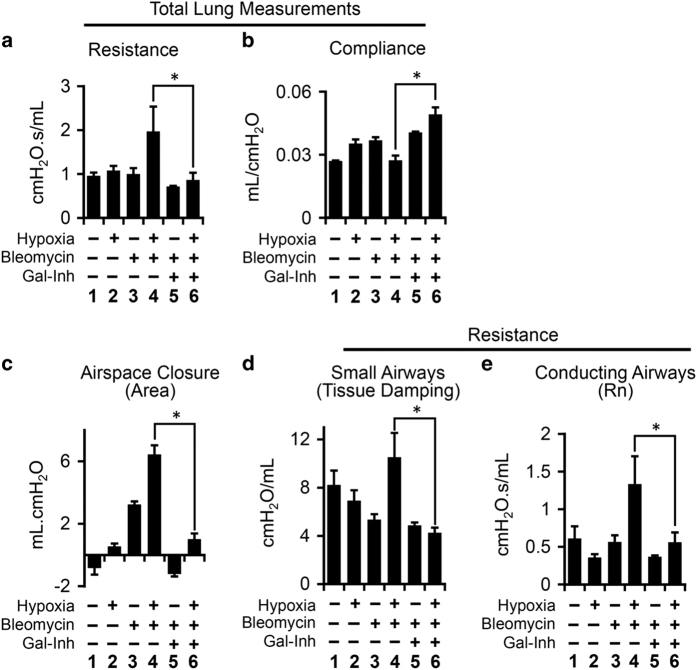
Idiopathic pulmonary fibrosis (IPF) is characterized by lung remodeling arising from epithelial injury, aberrant fibroblast growth, and excessive deposition of extracellular matrix. Repeated epithelial injury elicits abnormal wound repair and lung remodeling, often associated with alveolar collapse and edema, leading to focal hypoxia. Here, we demonstrate that hypoxia is a physiological insult that contributes to pulmonary fibrosis (PF) and define its molecular roles in profibrotic activation of lung epithelial cells. Hypoxia increased transcription of profibrotic genes and altered the proteomic signatures of lung epithelial cells. Network analysis of the hypoxic epithelial proteome revealed a crosstalk between transforming growth factor-1 and FAK1 (focal adhesion kinase-1) signaling, which regulated transcription of galectin-1, a profibrotic molecule. Galectin-1 physically interacted with and activated FAK1 in lung epithelial cells. We developed a novel model of exacerbated PF wherein hypoxia, as a secondary insult, caused PF in mice injured with subclinical levels of bleomycin. Hypoxia elevated expression of phosphorylated FAK1, galectin-1, and -smooth muscle actin and reduced caspase-3 activation, suggesting aberrant injury repair. Galectin-1 inhibition caused apoptosis in the lung parenchyma and reduced FAK1 activation, preventing the development of hypoxia-induced PF. Galectin-1 inhibition also attenuated fibrosis-associated lung function decline. Further, galectin-1 transcript levels were increased in the lungs of IPF patients. In summary, we have identified a profibrotic role of galectin-1 in hypoxia signaling driving PF.
特发性肺纤维化(IPF)的特征是由上皮损伤、异常的成纤维细胞生长和细胞外基质过度沉积引起的肺重塑。反复的上皮损伤引发异常的伤口修复和肺重塑,常伴有肺泡塌陷和水肿,导致局部缺氧。在此,我们证明缺氧是一种导致肺纤维化(PF)的生理损伤,并确定其在肺上皮细胞促纤维化激活中的分子作用。缺氧增加了促纤维化基因的转录,并改变了肺上皮细胞的蛋白质组特征。对缺氧上皮蛋白质组的网络分析揭示了转化生长因子-1和FAK1(粘着斑激酶-1)信号之间的相互作用,该相互作用调节了促纤维化分子半乳糖凝集素-1的转录。半乳糖凝集素-1在肺上皮细胞中与FAK1发生物理相互作用并激活FAK1。我们建立了一种新型的加重型PF模型,其中缺氧作为继发性损伤,在接受亚临床剂量博来霉素损伤的小鼠中导致PF。缺氧增加了磷酸化FAK1、半乳糖凝集素-1和α-平滑肌肌动蛋白的表达,并降低了caspase-3的激活,提示异常的损伤修复。抑制半乳糖凝集素-1可导致肺实质细胞凋亡并降低FAK1的激活,从而预防缺氧诱导的PF的发生。抑制半乳糖凝集素-1还可减轻与纤维化相关的肺功能下降。此外,IPF患者肺组织中半乳糖凝集素-1的转录水平升高。总之,我们已经确定了半乳糖凝集素-1在驱动PF的缺氧信号传导中的促纤维化作用。