Ando Maya, Fiesel Fabienne C, Hudec Roman, Caulfield Thomas R, Ogaki Kotaro, Górka-Skoczylas Paulina, Koziorowski Dariusz, Friedman Andrzej, Chen Li, Dawson Valina L, Dawson Ted M, Bu Guojun, Ross Owen A, Wszolek Zbigniew K, Springer Wolfdieter
Department of Neuroscience, Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL, 32224, USA.
Mayo Clinic Graduate School of Biomedical Sciences, Jacksonville, FL, 32224, USA.
Mol Neurodegener. 2017 Apr 24;12(1):32. doi: 10.1186/s13024-017-0174-z.
Mutations in PINK1 and PARKIN are the most common causes of recessive early-onset Parkinson's disease (EOPD). Together, the mitochondrial ubiquitin (Ub) kinase PINK1 and the cytosolic E3 Ub ligase PARKIN direct a complex regulated, sequential mitochondrial quality control. Thereby, damaged mitochondria are identified and targeted to degradation in order to prevent their accumulation and eventually cell death. Homozygous or compound heterozygous loss of either gene function disrupts this protective pathway, though at different steps and by distinct mechanisms. While structure and function of PARKIN variants have been well studied, PINK1 mutations remain poorly characterized, in particular under endogenous conditions. A better understanding of the exact molecular pathogenic mechanisms underlying the pathogenicity is crucial for rational drug design in the future.
Here, we characterized the pathogenicity of the PINK1 p.I368N mutation on the clinical and genetic as well as on the structural and functional level in patients' fibroblasts and in cell-based, biochemical assays.
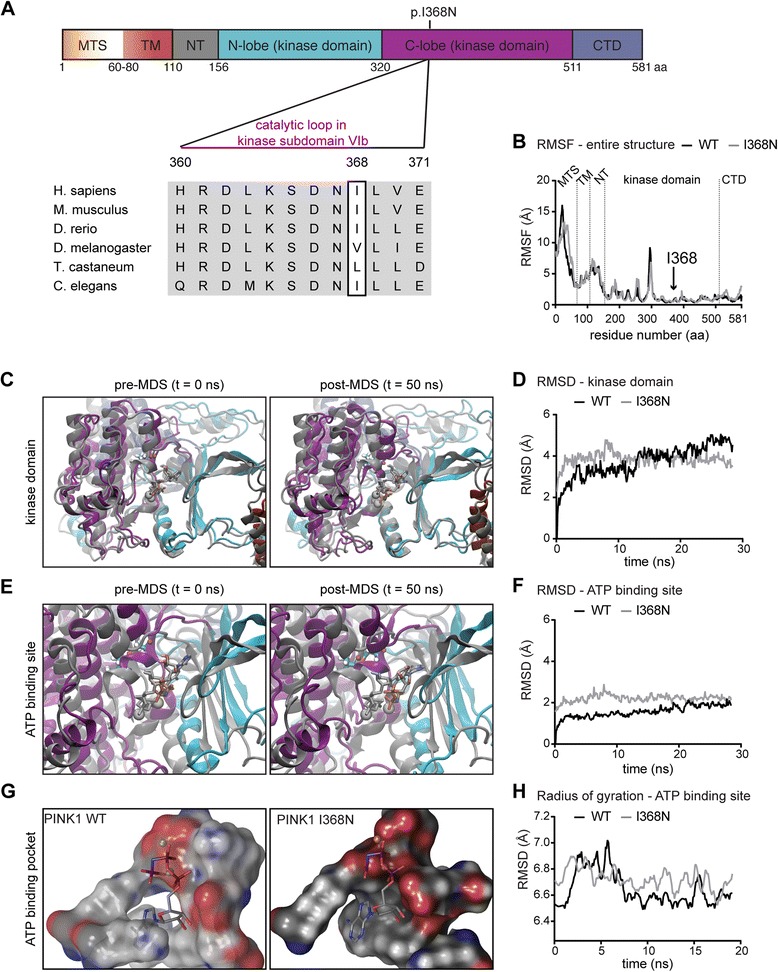
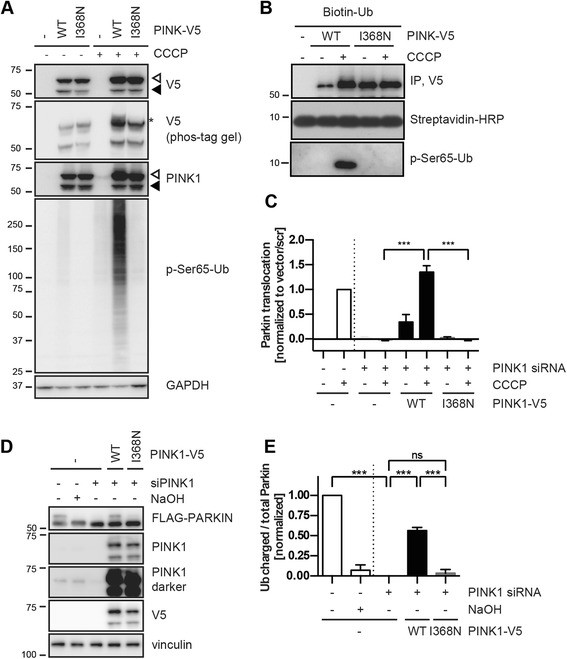
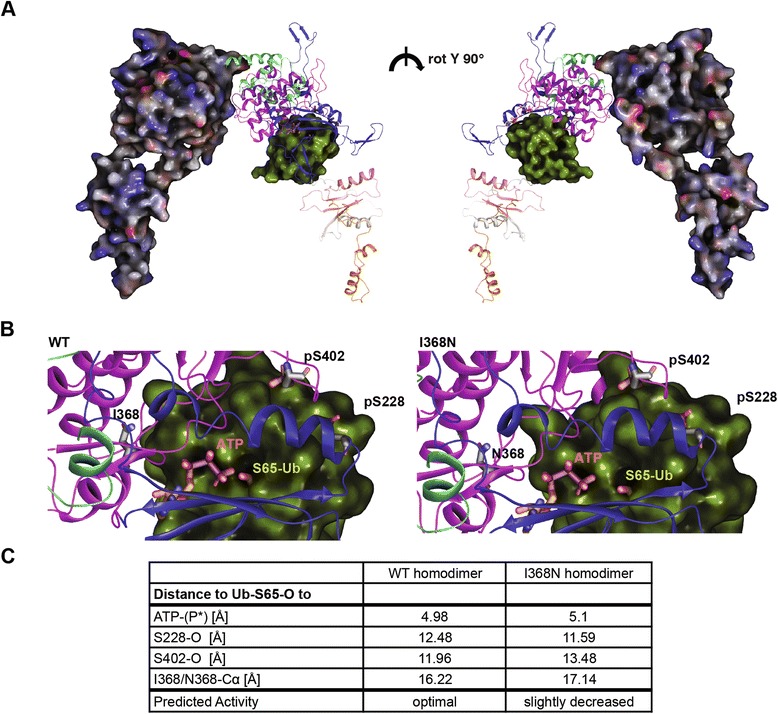
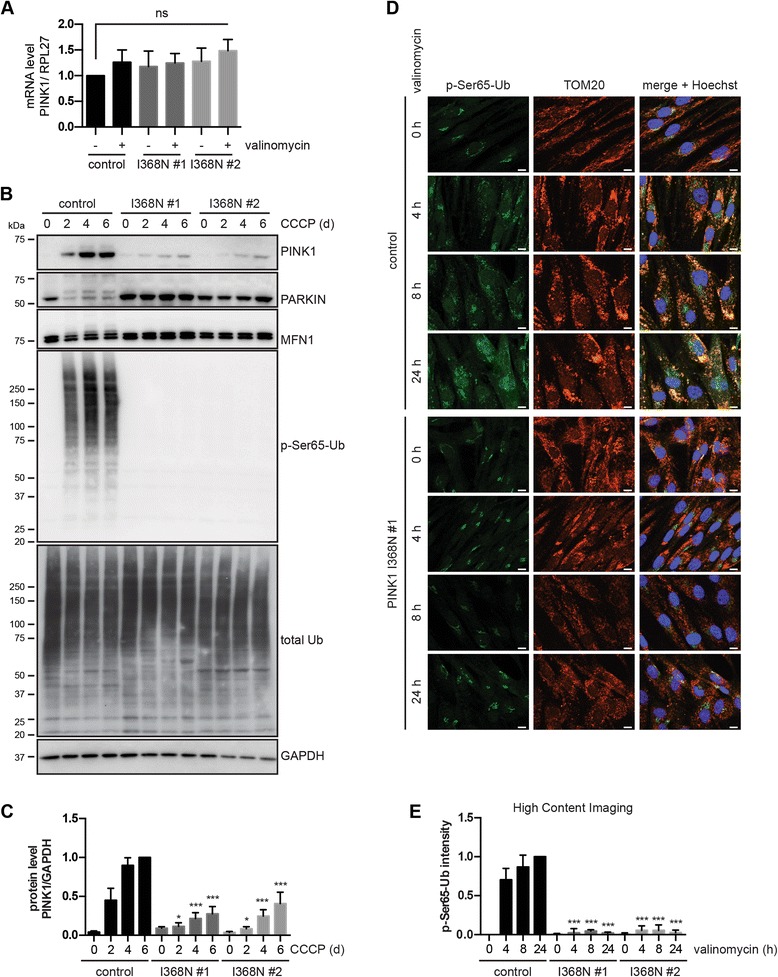
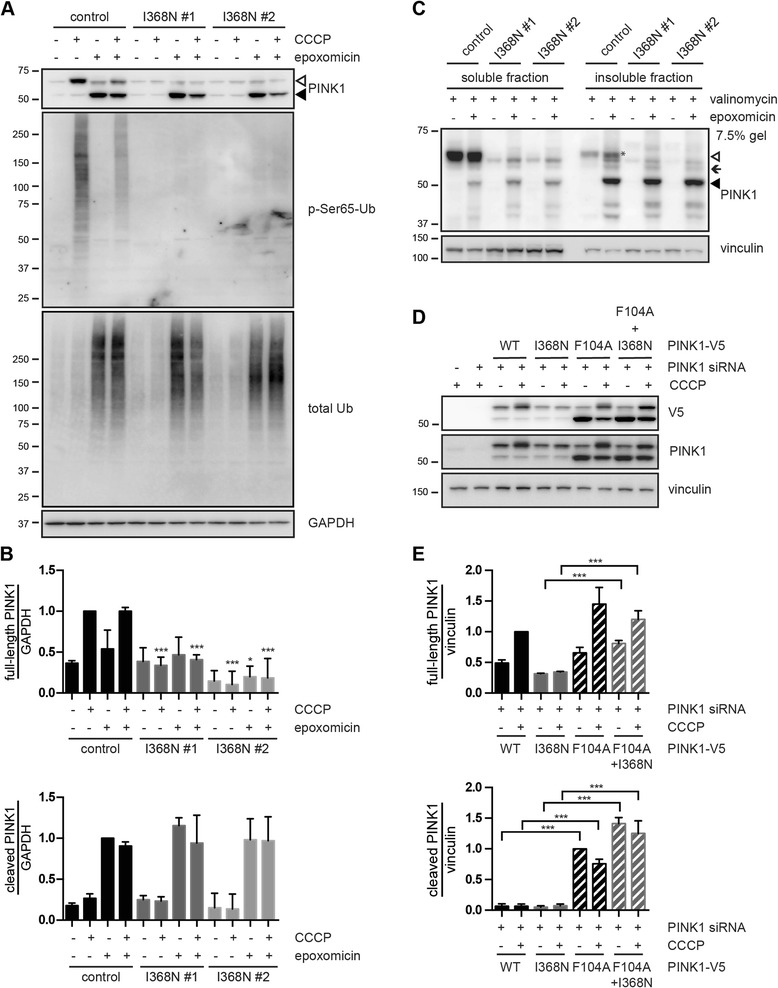
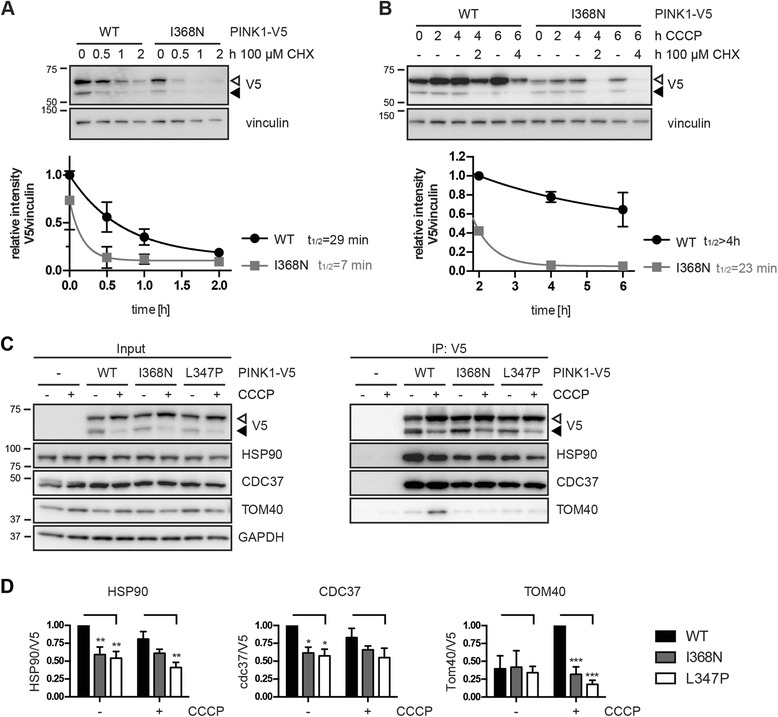
Under endogenous conditions, PINK1 p.I368N is expressed, imported, and N-terminally processed in healthy mitochondria similar to PINK1 wild type (WT). Upon mitochondrial damage, however, full-length PINK1 p.I368N is not sufficiently stabilized on the outer mitochondrial membrane (OMM) resulting in loss of mitochondrial quality control. We found that binding of PINK1 p.I368N to the co-chaperone complex HSP90/CDC37 is reduced and stress-induced interaction with TOM40 of the mitochondrial protein import machinery is abolished. Analysis of a structural PINK1 p.I368N model additionally suggested impairments of Ub kinase activity as the ATP-binding pocket was found deformed and the substrate Ub was slightly misaligned within the active site of the kinase. Functional assays confirmed the lack of Ub kinase activity.
Here we demonstrated that mutant PINK1 p.I368N can not be stabilized on the OMM upon mitochondrial stress and due to conformational changes in the active site does not exert kinase activity towards Ub. In patients' fibroblasts, biochemical assays and by structural analyses, we unraveled two pathomechanisms that lead to loss of function upon mutation of p.I368N and highlight potential strategies for future drug development.
PINK1和PARKIN基因的突变是隐性早发性帕金森病(EOPD)最常见的病因。线粒体泛素(Ub)激酶PINK1和胞质E3 Ub连接酶PARKIN共同指导一个复杂的、受调控的、连续的线粒体质量控制过程。由此,受损的线粒体被识别并靶向降解,以防止其积累并最终导致细胞死亡。任一基因功能的纯合或复合杂合缺失都会破坏这一保护途径,尽管在不同步骤和通过不同机制。虽然PARKIN变体的结构和功能已得到充分研究,但PINK1突变的特征仍然不明确,尤其是在内源条件下。更好地理解致病性的确切分子致病机制对于未来的合理药物设计至关重要。
在此,我们在患者成纤维细胞以及基于细胞的生化分析中,从临床、遗传以及结构和功能水平上对PINK1 p.I368N突变的致病性进行了表征。
在内源条件下,PINK1 p.I368N在健康线粒体中表达、导入并进行N端加工,类似于PINK1野生型(WT)。然而,在线粒体损伤时,全长PINK1 p.I368N在外膜线粒体(OMM)上不能充分稳定,导致线粒体质量控制丧失。我们发现PINK1 p.I368N与伴侣蛋白复合物HSP90/CDC37的结合减少,并且应激诱导的与线粒体蛋白导入机制的TOM40的相互作用被消除。对PINK1 p.I368N结构模型的分析还表明,由于ATP结合口袋变形且底物Ub在激酶活性位点内略有错位,泛素激酶活性受损。功能分析证实缺乏泛素激酶活性。
在此我们证明,突变型PINK1 p.I368N在线粒体应激时不能在OMM上稳定,并且由于活性位点的构象变化,对Ub不发挥激酶活性。在患者成纤维细胞、生化分析和结构分析中,我们揭示了两种致病机制,这些机制导致p.I368N突变后功能丧失,并突出了未来药物开发的潜在策略。