Chatzispyrou Iliana A, Alders Marielle, Guerrero-Castillo Sergio, Zapata Perez Ruben, Haagmans Martin A, Mouchiroud Laurent, Koster Janet, Ofman Rob, Baas Frank, Waterham Hans R, Spelbrink Johannes N, Auwerx Johan, Mannens Marcel M, Houtkooper Riekelt H, Plomp Astrid S
Laboratory Genetic Metabolic Diseases, Academic Medical Center, 1105 AZ Amsterdam, The Netherlands.
Department of Clinical Genetics, Academic Medical Center, 1105 AZ Amsterdam, The Netherlands.
Hum Mol Genet. 2017 Jul 1;26(13):2541-2550. doi: 10.1093/hmg/ddx152.
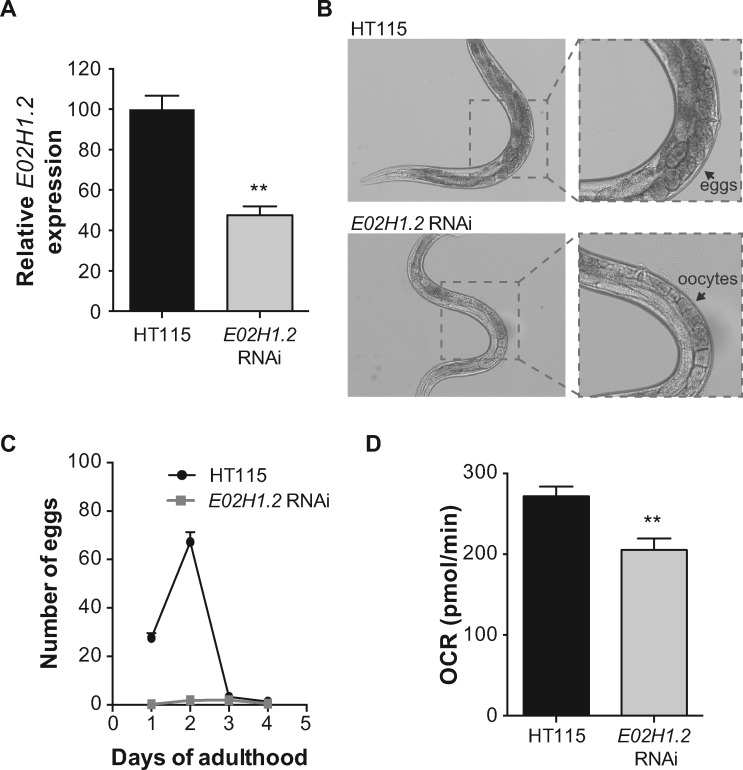
Perrault syndrome (PS) is a rare recessive disorder characterized by ovarian dysgenesis and sensorineural deafness. It is clinically and genetically heterogeneous, and previously mutations have been described in different genes, mostly related to mitochondrial proteostasis. We diagnosed three unrelated females with PS and set out to identify the underlying genetic cause using exome sequencing. We excluded mutations in the known PS genes, but identified a single homozygous mutation in the ERAL1 gene (c.707A > T; p.Asn236Ile). Since ERAL1 protein binds to the mitochondrial 12S rRNA and is involved in the assembly of the small mitochondrial ribosomal subunit, the identified variant represented a likely candidate. In silico analysis of a 3D model for ERAL1 suggested that the mutated residue hinders protein-substrate interactions, potentially affecting its function. On a molecular basis, PS skin fibroblasts had reduced ERAL1 protein levels. Complexome profiling of the cells showed an overall decrease in the levels of assembled small ribosomal subunit, indicating that the ERAL1 variant affects mitochondrial ribosome assembly. Moreover, levels of the 12S rRNA were reduced in the patients, and were rescued by lentiviral expression of wild type ERAL1. At the physiological level, mitochondrial respiration was markedly decreased in PS fibroblasts, confirming disturbed mitochondrial function. Finally, knockdown of the C. elegans ERAL1 homologue E02H1.2 almost completely blocked egg production in worms, mimicking the compromised fertility in PS-affected women. Our cross-species data in patient cells and worms support the hypothesis that mutations in ERAL1 can cause PS and are associated with changes in mitochondrial metabolism.
佩罗特综合征(PS)是一种罕见的隐性疾病,其特征为卵巢发育不全和感音神经性耳聋。它在临床和遗传方面具有异质性,此前已在不同基因中发现了突变,这些基因大多与线粒体蛋白质稳态相关。我们诊断出三名无血缘关系的患有PS的女性,并着手使用外显子组测序来确定潜在的遗传病因。我们排除了已知PS基因中的突变,但在ERAL1基因中鉴定出一个纯合突变(c.707A>T;p.Asn236Ile)。由于ERAL1蛋白与线粒体12S rRNA结合并参与小线粒体核糖体亚基的组装,因此鉴定出的变体可能是一个候选基因。对ERAL1的三维模型进行的计算机模拟分析表明,突变残基阻碍了蛋白质与底物的相互作用,可能会影响其功能。在分子水平上,PS皮肤成纤维细胞中ERAL1蛋白水平降低。对细胞进行的复合物组分析显示,组装好的小核糖体亚基水平总体下降,表明ERAL1变体影响线粒体核糖体的组装。此外,患者体内12S rRNA水平降低,通过慢病毒表达野生型ERAL1可使其恢复。在生理水平上,PS成纤维细胞中的线粒体呼吸明显降低,证实了线粒体功能受到干扰。最后,敲低秀丽隐杆线虫ERAL1的同源物E02H1.2几乎完全阻断了线虫的产卵,这与PS患者受损的生育能力相似。我们在患者细胞和线虫中的跨物种数据支持了这样的假设,即ERAL1突变可导致PS,并与线粒体代谢变化相关。