Smith Thomas B, Kopajtich Robert, Demain Leigh A M, Rea Alessandro, Thomas Huw B, Schiff Manuel, Beetz Christian, Joss Shelagh, Conway Gerard S, Shukla Anju, Yeole Mayuri, Radhakrishnan Periyasamy, Azzouz Hatem, Ben Chehida Amel, Elmaleh-Bergès Monique, Glasgow Ruth I C, Thompson Kyle, Oláhová Monika, He Langping, Jenkinson Emma M, Jahic Amir, Belyantseva Inna A, Barzik Melanie, Urquhart Jill E, O' Sullivan James, Williams Simon G, Bhaskar Sanjeev S, Carrera Samantha, Blakes Alexander J M, Banka Siddharth, Yue Wyatt W, Ellingford Jamie M, Houlden Henry, Munro Kevin J, Friedman Thomas B, Taylor Robert W, Prokisch Holger, O'Keefe Raymond T, Newman William G
Division of Evolution, Infection and Genomics, School of Biological Sciences, The University of Manchester, Manchester, M13 9PL, UK.
Manchester Centre for Genomic Medicine, St Mary's Hospital, The University of Manchester NHS Foundation Trust, Manchester, M13 9WL, UK.
medRxiv. 2024 Aug 21:2024.08.19.24312079. doi: 10.1101/2024.08.19.24312079.
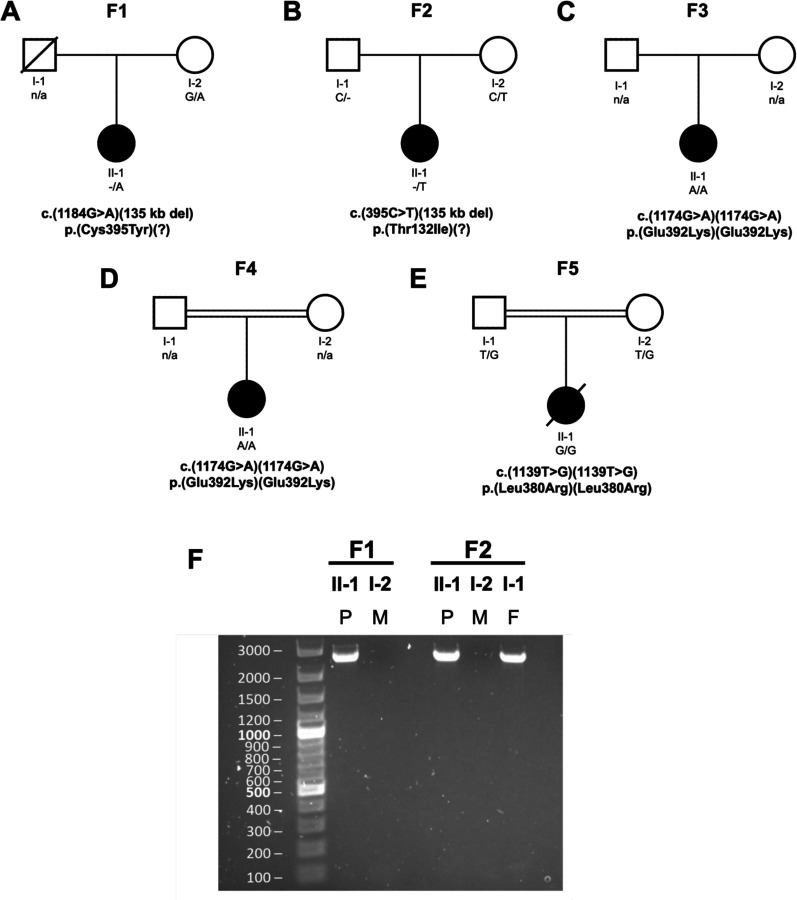
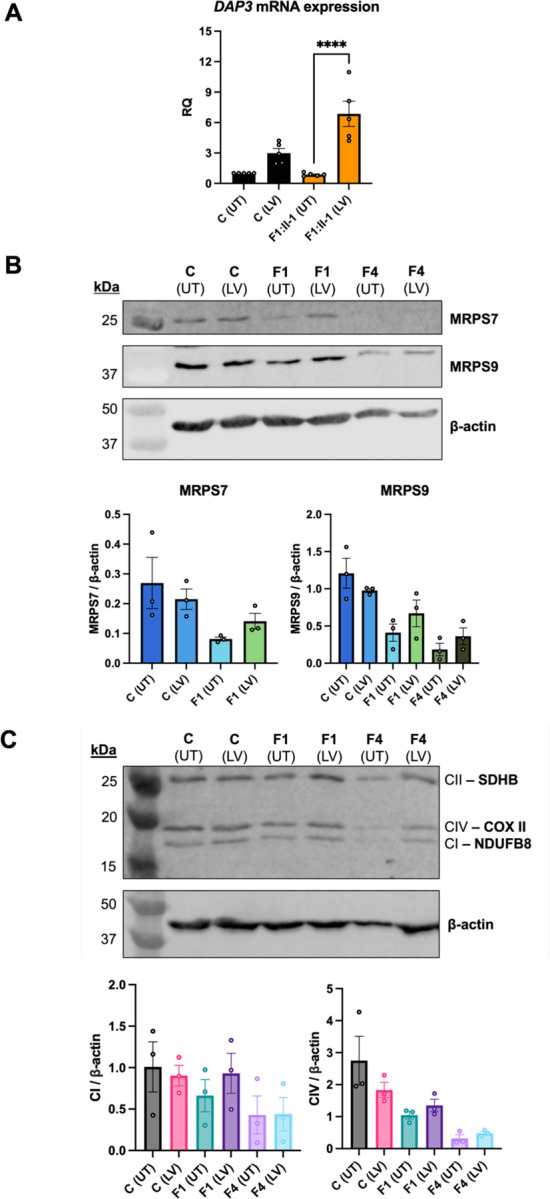
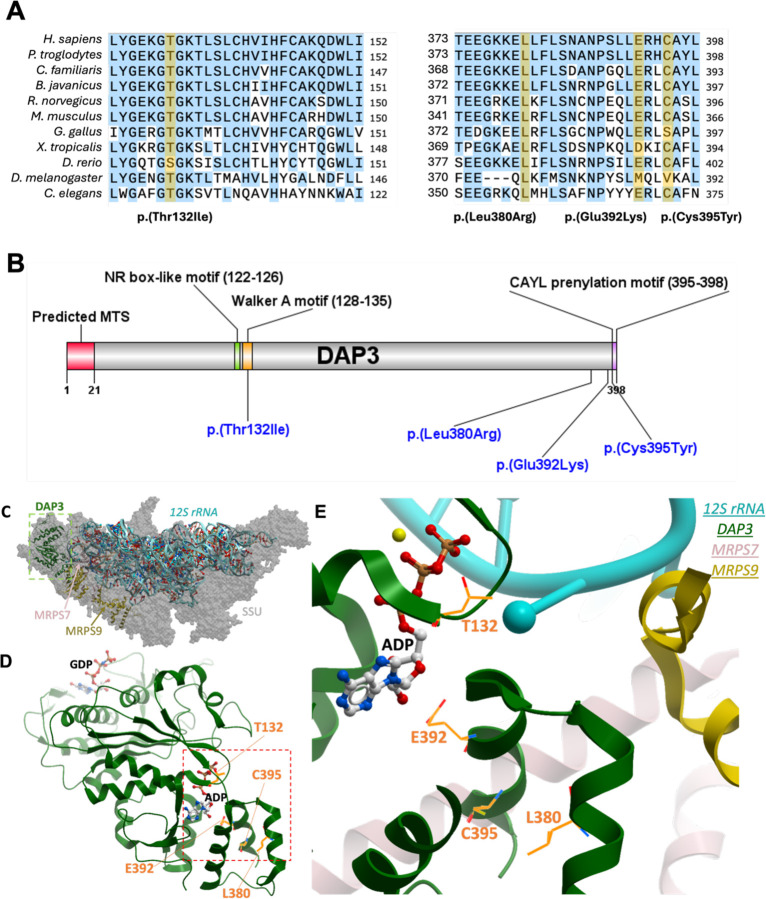
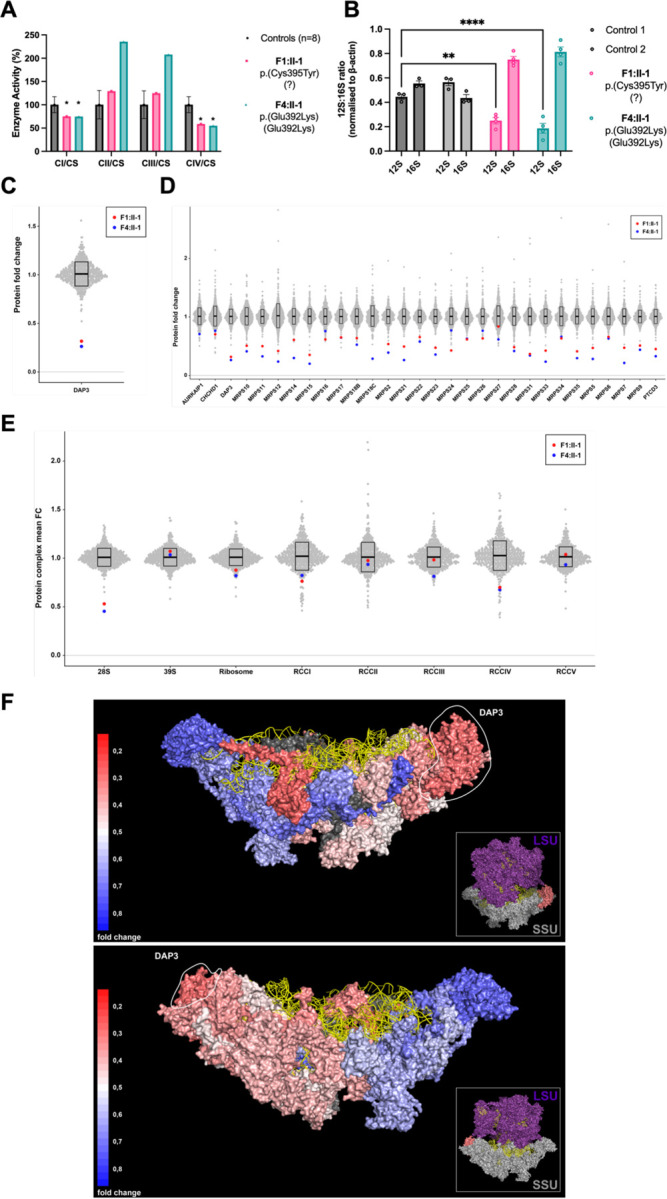
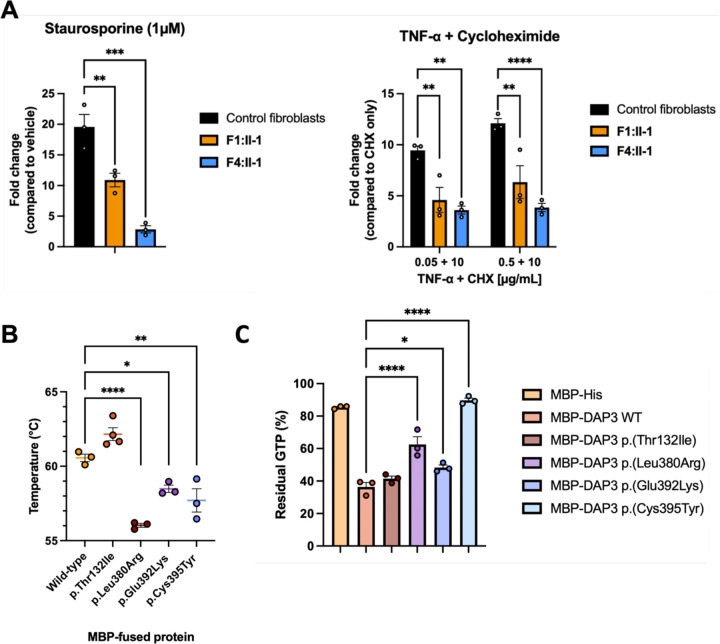
The mitoribosome synthesizes 13 protein subunits of the oxidative phosphorylation system encoded by the mitochondrial genome. The mitoribosome is composed of 12S rRNA, 16S rRNA and 82 mitoribosomal proteins encoded by nuclear genes. To date, variants in 12 genes encoding mitoribosomal proteins are associated with rare monogenic disorders, and frequently show combined oxidative phosphorylation deficiency. Here, we describe five unrelated individuals with biallelic variants in the nuclear gene encoding mitoribosomal small subunit 29 (MRPS29), with variable clinical presentations ranging from Perrault syndrome (sensorineural hearing loss and ovarian insufficiency) to an early childhood neurometabolic phenotype. Assessment of respiratory chain function and proteomic profiling of fibroblasts from affected individuals demonstrated reduced MRPS29 protein levels, and consequently decreased levels of additional protein components of the mitoribosomal small subunit, associated with a combined complex I and IV deficiency. Lentiviral transduction of fibroblasts from affected individuals with wild-type cDNA increased DAP3 mRNA expression, and partially rescued protein levels of MRPS7, MRPS9 and complex I and IV subunits, demonstrating the pathogenicity of the variants. Protein modelling suggested that disease-associated missense variants can impact ADP binding, and assays demonstrated variants can consequently reduce both intrinsic and extrinsic apoptotic sensitivity, DAP3 thermal stability and DAP3 GTPase activity. Our study presents genetic and functional evidence that biallelic variants in result in a multisystem disorder of combined oxidative phosphorylation deficiency with pleiotropic presentations, consistent with mitochondrial dysfunction.
线粒体核糖体合成由线粒体基因组编码的氧化磷酸化系统的13种蛋白质亚基。线粒体核糖体由12S rRNA、16S rRNA和由核基因编码的82种线粒体核糖体蛋白组成。迄今为止,编码线粒体核糖体蛋白的12个基因中的变异与罕见的单基因疾病相关,并且经常表现出联合氧化磷酸化缺陷。在这里,我们描述了5名无关个体,他们在编码线粒体核糖体小亚基29(MRPS29)的核基因中存在双等位基因变异,临床表现各异,从佩罗综合征(感音神经性听力损失和卵巢功能不全)到幼儿期神经代谢表型。对受影响个体的成纤维细胞进行呼吸链功能评估和蛋白质组分析,结果显示MRPS29蛋白水平降低,因此线粒体核糖体小亚基的其他蛋白质成分水平也降低,这与复合体I和IV联合缺陷有关。用野生型cDNA对受影响个体的成纤维细胞进行慢病毒转导可增加DAP3 mRNA表达,并部分挽救MRPS7、MRPS9以及复合体I和IV亚基的蛋白质水平,证明了这些变异的致病性。蛋白质建模表明,与疾病相关的错义变异会影响ADP结合,并且实验表明这些变异会因此降低内在和外在的凋亡敏感性、DAP3热稳定性和DAP3 GTPase活性。我们的研究提供了遗传和功能证据,表明双等位基因变异导致了一种多系统疾病,表现为联合氧化磷酸化缺陷和多效性表现,与线粒体功能障碍一致。