White Jessica R, Gong Huiyu, Pope Brock, Schlievert Patrick, McElroy Steven J
Pediatrics, University of Iowa, Iowa City, IA 54424, USA.
Microbiology, University of Iowa, Iowa City, IA 54424, USA.
Dis Model Mech. 2017 Jun 1;10(6):727-736. doi: 10.1242/dmm.028589. Epub 2017 Apr 27.
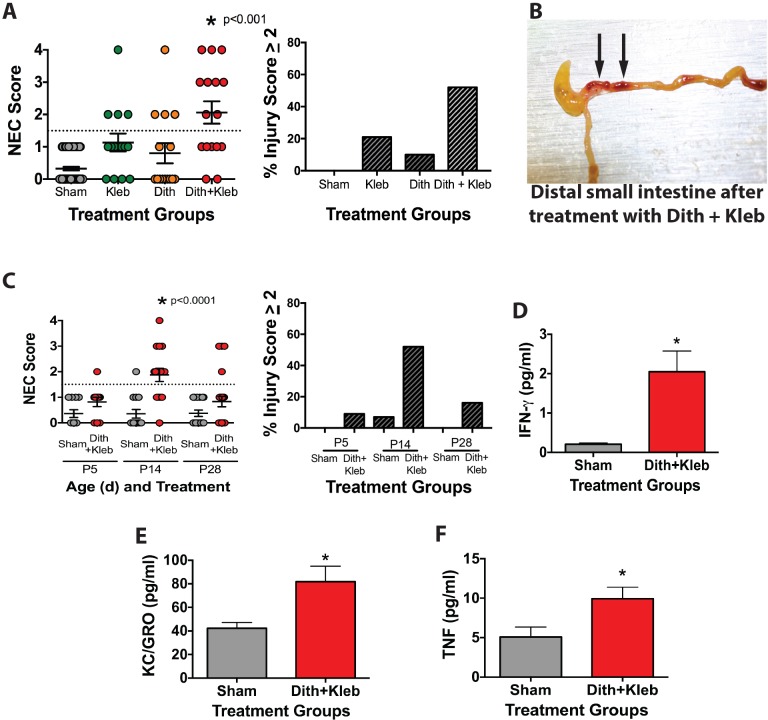
Necrotizing enterocolitis (NEC) remains a leading cause of morbidity and mortality in premature infants. Both human surgical specimens and animal models suggest a potential involvement of Paneth cells in NEC pathogenesis. Paneth cells play critical roles in epithelial homeostasis, innate immunity and host-microbial interactions. Yet, the complex interplay between Paneth cell disruption, epithelial barrier dysfunction and microbial-driven inflammation remains unclear in the immature intestine. In this study, mucosal intestinal injury consistent with human NEC was induced in postnatal day 14-16 (P14-P16) mice by disrupting Paneth cells, followed by gavage with Mucosal injury was determined by histology, serum cytokine levels and epithelial barrier dysfunction. Toll-like receptor 4 (TLR4) activation was examined using protein expression, gene expression, and mice. Finally, the role of bacteria was evaluated using heat-killed bacteria, conditioned media, and cecal slurries. We found that live bacteria were required to induce injury; however, TLR4 activation was not required. NEC induced by Paneth cell disruption results in altered localization of tight junction proteins and subsequent loss of barrier function. Prior research has shown a requirement for TLR4 activation to induce NEC-like damage. However, many infants develop NEC in the absence of Gram-negative rod bacteremia, raising the possibility that alternative pathways to intestinal injury exist. In this study, we show a previously unknown mechanism for the development of intestinal injury equivalent to that seen in human NEC and that is not dependent on TLR4 pathways. These data are congruent with the new hypothesis that NEC may be the consequence of several disease processes ending in a final common inflammatory pathway.
坏死性小肠结肠炎(NEC)仍然是早产儿发病和死亡的主要原因。人体手术标本和动物模型均提示潘氏细胞可能参与NEC的发病机制。潘氏细胞在上皮内环境稳定、固有免疫及宿主与微生物的相互作用中发挥关键作用。然而,在未成熟肠道中,潘氏细胞破坏、上皮屏障功能障碍和微生物驱动的炎症之间复杂的相互作用仍不清楚。在本研究中,通过破坏出生后第14 - 16天(P14 - P16)小鼠的潘氏细胞,诱导出与人类NEC一致的肠道黏膜损伤,随后经口灌胃……通过组织学、血清细胞因子水平及上皮屏障功能障碍来确定黏膜损伤情况。使用蛋白质表达、基因表达及……检测Toll样受体4(TLR4)的激活情况。最后,使用热灭活细菌、条件培养基及盲肠匀浆评估细菌的作用。我们发现诱导损伤需要活菌;然而,并不需要TLR4激活。潘氏细胞破坏诱导的NEC导致紧密连接蛋白定位改变及随后的屏障功能丧失。先前的研究表明诱导NEC样损伤需要TLR4激活。然而,许多婴儿在无革兰氏阴性杆菌血症的情况下发生NEC,这增加了存在肠道损伤替代途径的可能性。在本研究中,我们展示了一种此前未知的、与人类NEC所见类似的肠道损伤发生机制,且该机制不依赖于TLR4途径。这些数据与新的假说一致,即NEC可能是几种疾病过程最终导致共同炎症途径的结果。