Chalise Prabhakar, Fridley Brooke L
Department of Biostatistics, University of Kansas Medical Center, Kansas City, Kansas, United States of America.
Department of Biostatistics and Bioinformatics, Moffitt Cancer Center, Tampa, Florida, United States of America.
PLoS One. 2017 May 1;12(5):e0176278. doi: 10.1371/journal.pone.0176278. eCollection 2017.
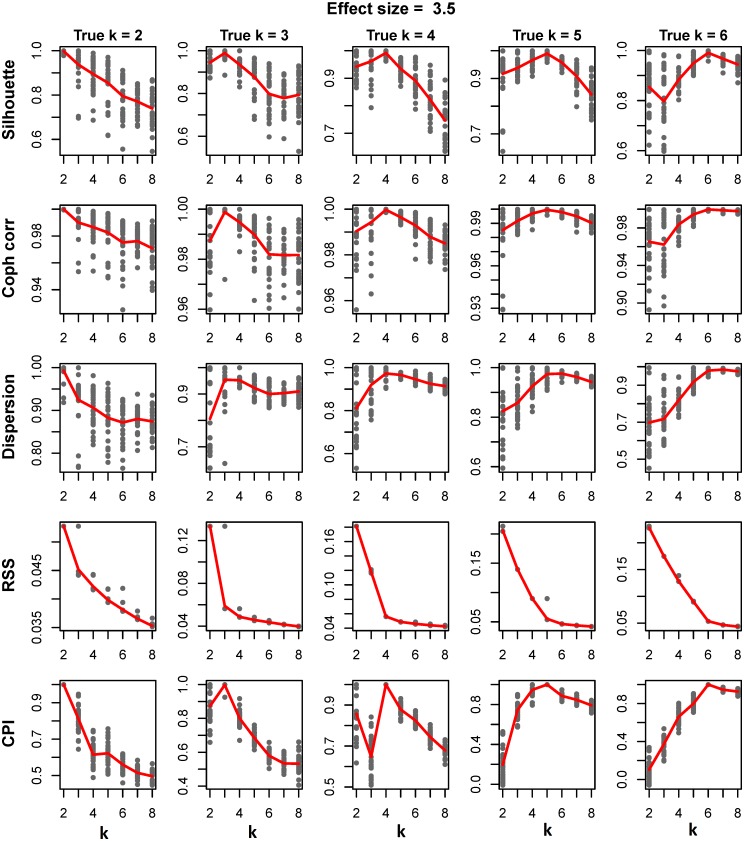
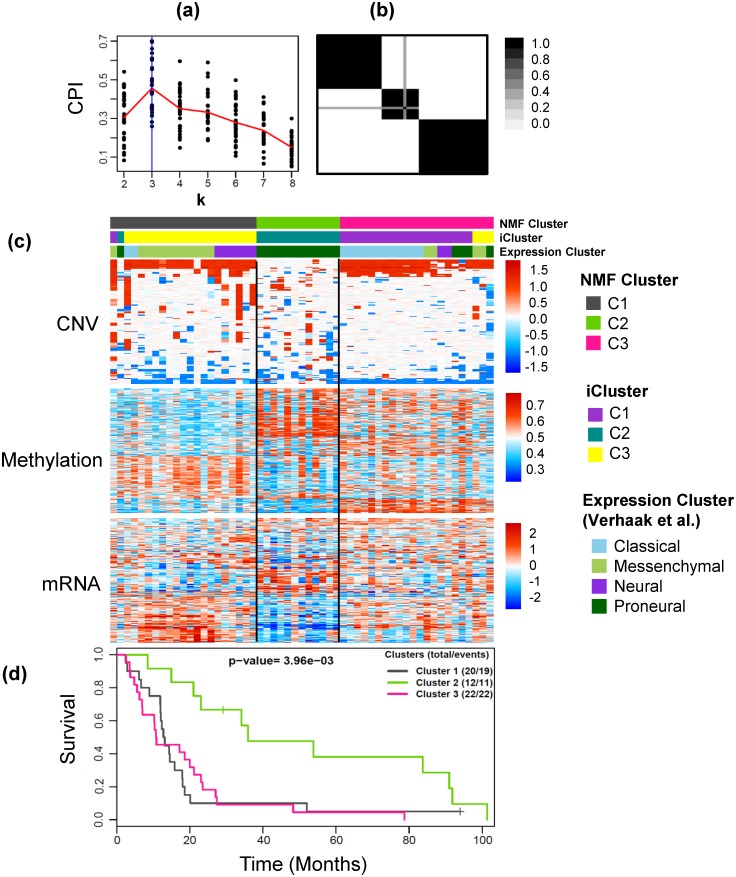
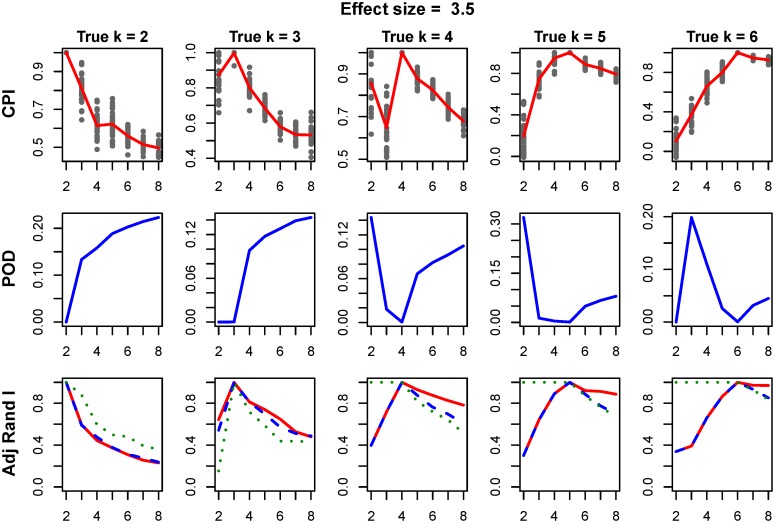
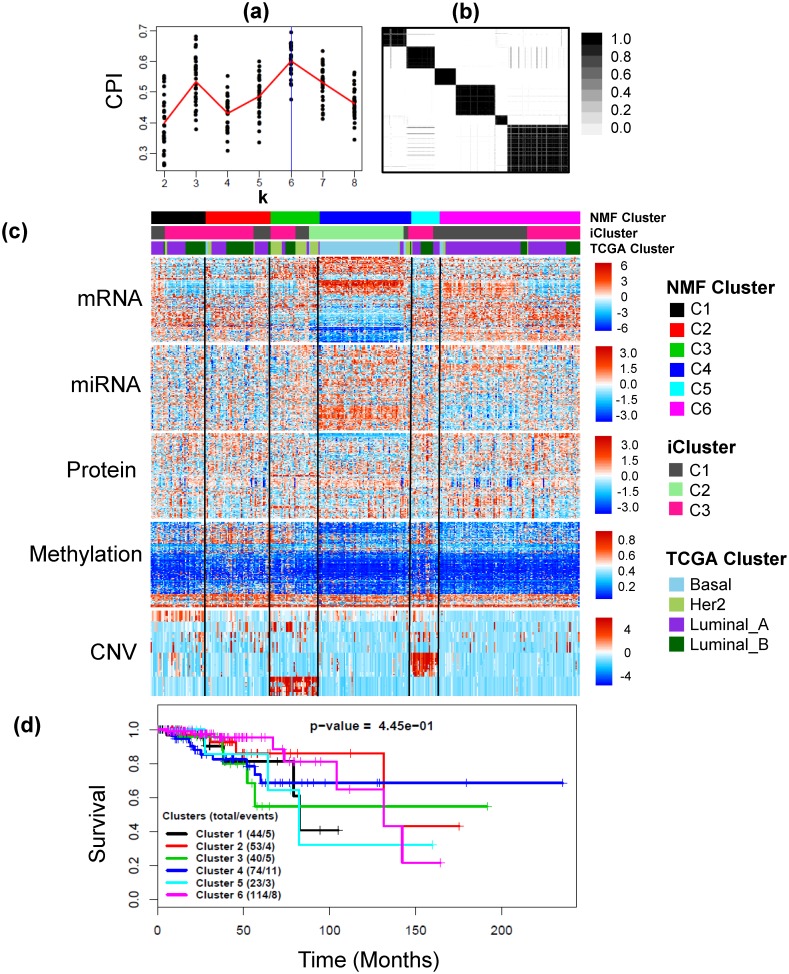
Integrative analyses of high-throughput 'omic data, such as DNA methylation, DNA copy number alteration, mRNA and protein expression levels, have created unprecedented opportunities to understand the molecular basis of human disease. In particular, integrative analyses have been the cornerstone in the study of cancer to determine molecular subtypes within a given cancer. As malignant tumors with similar morphological characteristics have been shown to exhibit entirely different molecular profiles, there has been significant interest in using multiple 'omic data for the identification of novel molecular subtypes of disease, which could impact treatment decisions. Therefore, we have developed intNMF, an integrative approach for disease subtype classification based on non-negative matrix factorization. The proposed approach carries out integrative clustering of multiple high dimensional molecular data in a single comprehensive analysis utilizing the information across multiple biological levels assessed on the same individual. As intNMF does not assume any distributional form for the data, it has obvious advantages over other model based clustering methods which require specific distributional assumptions. Application of intNMF is illustrated using both simulated and real data from The Cancer Genome Atlas (TCGA).
对高通量“组学”数据(如DNA甲基化、DNA拷贝数改变、mRNA和蛋白质表达水平)进行综合分析,为理解人类疾病的分子基础创造了前所未有的机会。特别是,综合分析一直是癌症研究中确定特定癌症分子亚型的基石。由于具有相似形态特征的恶性肿瘤已被证明表现出完全不同的分子谱,因此人们对使用多种“组学”数据来识别疾病的新型分子亚型产生了浓厚兴趣,这可能会影响治疗决策。因此,我们开发了intNMF,一种基于非负矩阵分解的疾病亚型分类综合方法。该方法在单一综合分析中对多个高维分子数据进行综合聚类,利用在同一个体上评估的多个生物学水平的信息。由于intNMF不假设数据的任何分布形式,与其他需要特定分布假设的基于模型的聚类方法相比,它具有明显优势。使用来自癌症基因组图谱(TCGA)的模拟数据和真实数据说明了intNMF的应用。