Hinze S J, Jackson M R, Lie S, Jolly L, Field M, Barry S C, Harvey R J, Shoubridge C
Department of Paediatrics, Adelaide School of Medicine, University of Adelaide, Adelaide, SA, Australia.
Robinson Research Institute, University of Adelaide, Adelaide, SA, Australia.
Transl Psychiatry. 2017 May 2;7(5):e1110. doi: 10.1038/tp.2017.81.
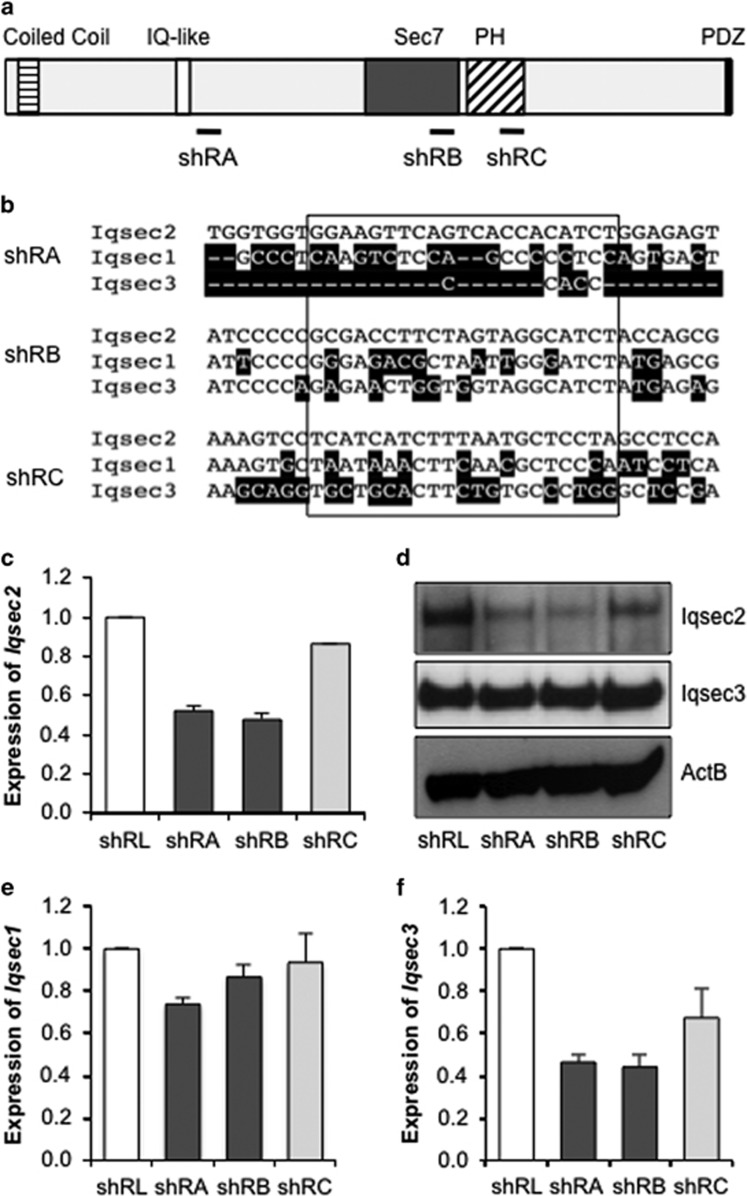
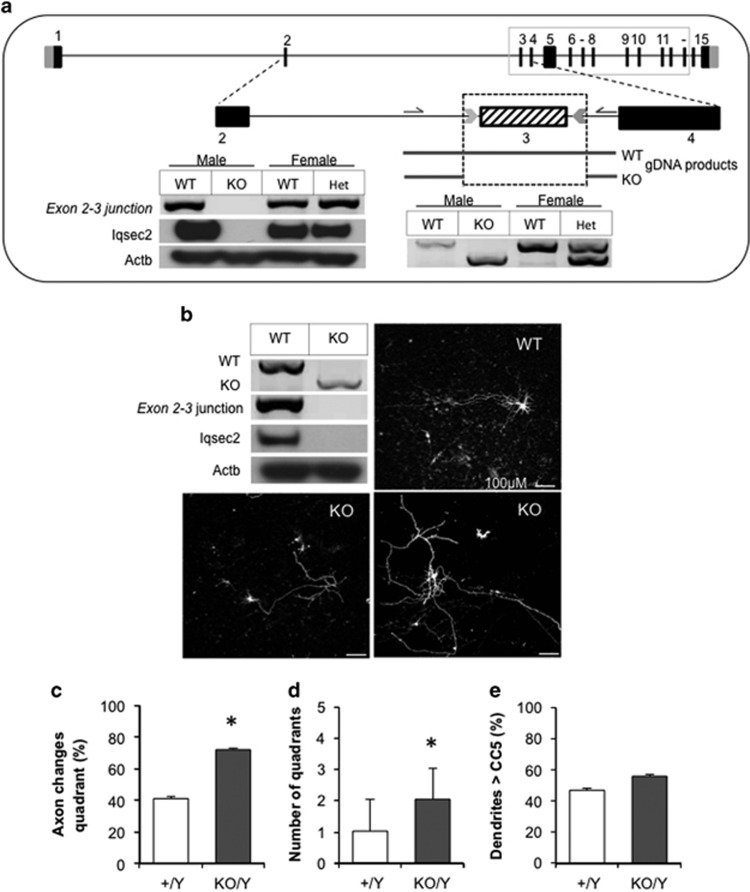
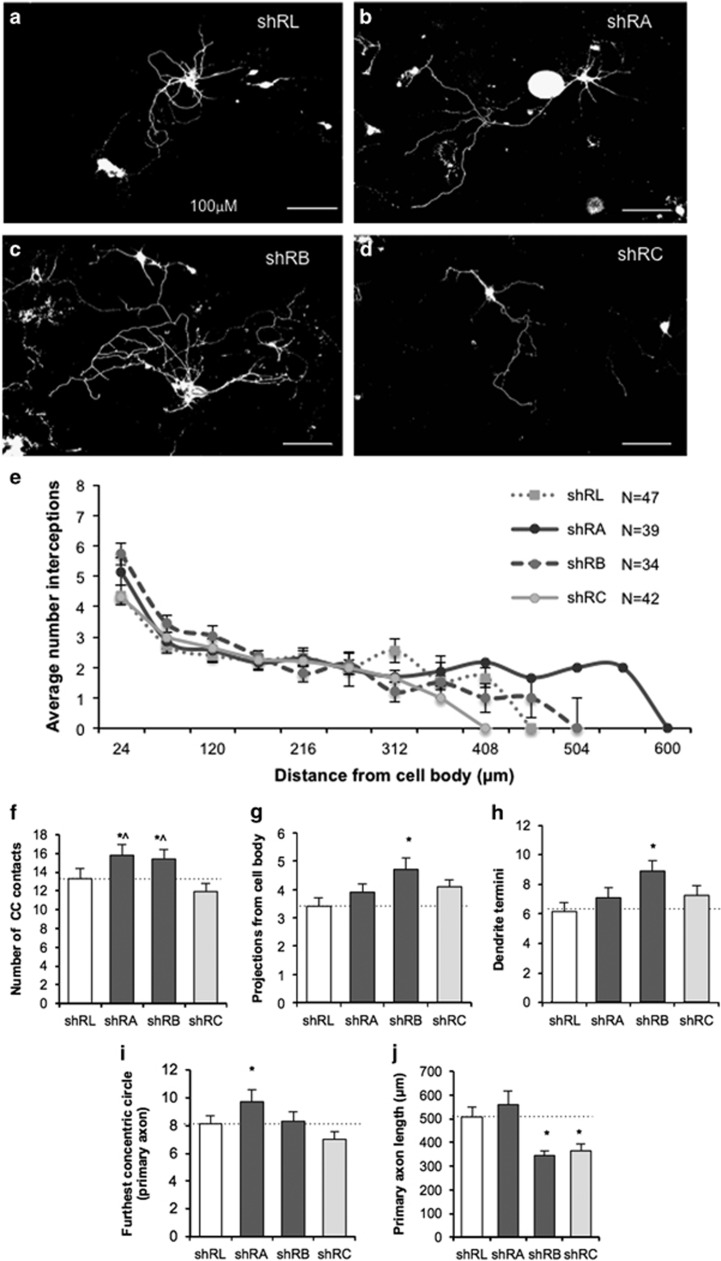
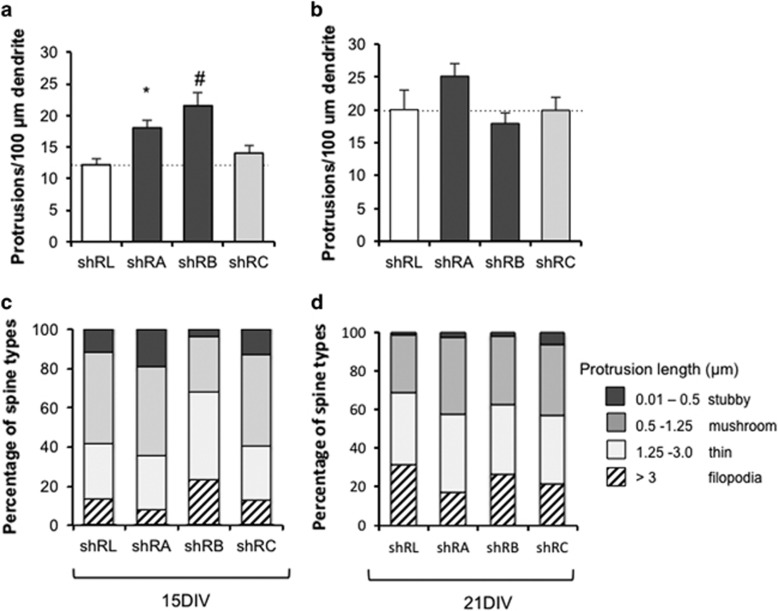
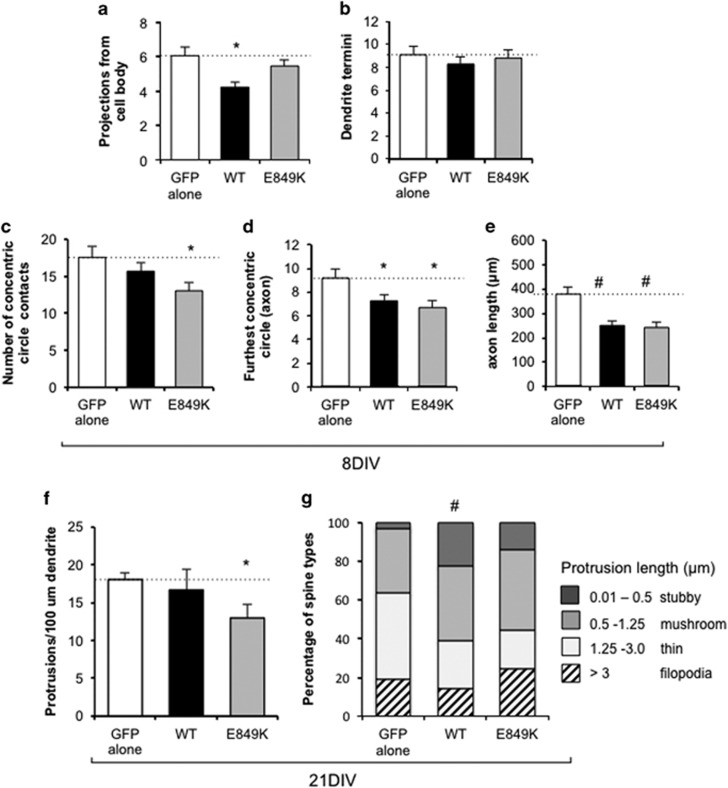
There is considerable genetic and phenotypic heterogeneity associated with intellectual disability (ID), specific learning disabilities, attention-deficit hyperactivity disorder, autism and epilepsy. The intelligence quotient (IQ) motif and SEC7 domain containing protein 2 gene (IQSEC2) is located on the X-chromosome and harbors mutations that contribute to non-syndromic ID with and without early-onset seizure phenotypes in both sexes. Although IQ and Sec7 domain mutations lead to partial loss of IQSEC2 enzymatic activity, the in vivo pathogenesis resulting from these mutations is not known. Here we reveal that IQSEC2 has a key role in dendritic spine morphology. Partial loss-of-function mutations were modeled using a lentiviral short hairpin RNA (shRNA) approach, which achieved a 57% knockdown of Iqsec2 expression in primary hippocampal cell cultures from mice. Investigating gross morphological parameters after 8 days of in vitro culture (8DIV) identified a 32% reduction in primary axon length, in contrast to a 27% and 31% increase in the number and complexity of dendrites protruding from the cell body, respectively. This increase in dendritic complexity and spread was carried through dendritic spine development, with a 34% increase in the number of protrusions per dendritic segment compared with controls at 15DIV. Although the number of dendritic spines had normalized by 21DIV, a reduction was noted in the number of immature spines. In contrast, when modeling increased dosage, overexpression of wild-type IQSEC2 led to neurons with shorter axons that were more compact and displayed simpler dendritic branching. Disturbances to dendritic morphology due to knockdown of Iqsec2 were recapitulated in neurons from Iqsec2 knockout mice generated in our laboratory using CRISPR/Cas9 technology. These observations provide evidence of dosage sensitivity for IQSEC2, which normally escapes X-inactivation in females, and links these disturbances in expression to alterations in the morphology of developing neurons.
智力残疾(ID)、特定学习障碍、注意力缺陷多动障碍、自闭症和癫痫都存在相当大的遗传和表型异质性。含智力商数(IQ)基序和SEC7结构域蛋白2基因(IQSEC2)位于X染色体上,其携带的突变会导致男女出现有无早发性癫痫表型的非综合征性ID。尽管IQ和Sec7结构域突变会导致IQSEC2酶活性部分丧失,但这些突变导致的体内发病机制尚不清楚。在此,我们揭示IQSEC2在树突棘形态中起关键作用。使用慢病毒短发夹RNA(shRNA)方法模拟功能部分丧失突变,该方法在小鼠原代海马细胞培养物中实现了Iqsec2表达57%的敲低。在体外培养8天(8DIV)后研究总体形态参数发现,初级轴突长度减少了32%,相比之下,从细胞体伸出的树突数量和复杂性分别增加了27%和31%。这种树突复杂性和扩展的增加贯穿树突棘发育过程,与15DIV时的对照组相比,每个树突段的突起数量增加了34%。尽管到21DIV时树突棘数量已恢复正常,但未成熟棘突的数量有所减少。相反,在模拟剂量增加时,野生型IQSEC2的过表达导致神经元轴突更短、更紧凑且树突分支更简单。在我们实验室使用CRISPR/Cas9技术生成的Iqsec2基因敲除小鼠的神经元中,也再现了因Iqsec2敲低导致的树突形态紊乱。这些观察结果提供了IQSEC2剂量敏感性的证据,IQSEC2在雌性中通常逃避X染色体失活,并将这些表达紊乱与发育中神经元形态的改变联系起来。