Geraets Ryan D, Langin Logan M, Cain Jacob T, Parker Camille M, Beraldi Rosanna, Kovacs Attila D, Weimer Jill M, Pearce David A
Children's Health Research Center, Sanford Research, Sioux Falls, South Dakota, United States of America.
Sanford School of Medicine at the University of South Dakota, Sioux Falls, South Dakota, United States of America.
PLoS One. 2017 May 2;12(5):e0176526. doi: 10.1371/journal.pone.0176526. eCollection 2017.
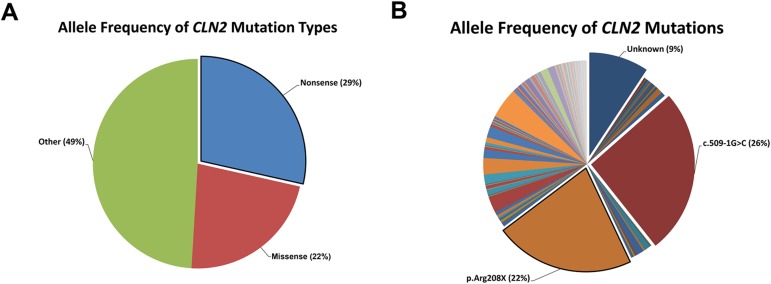
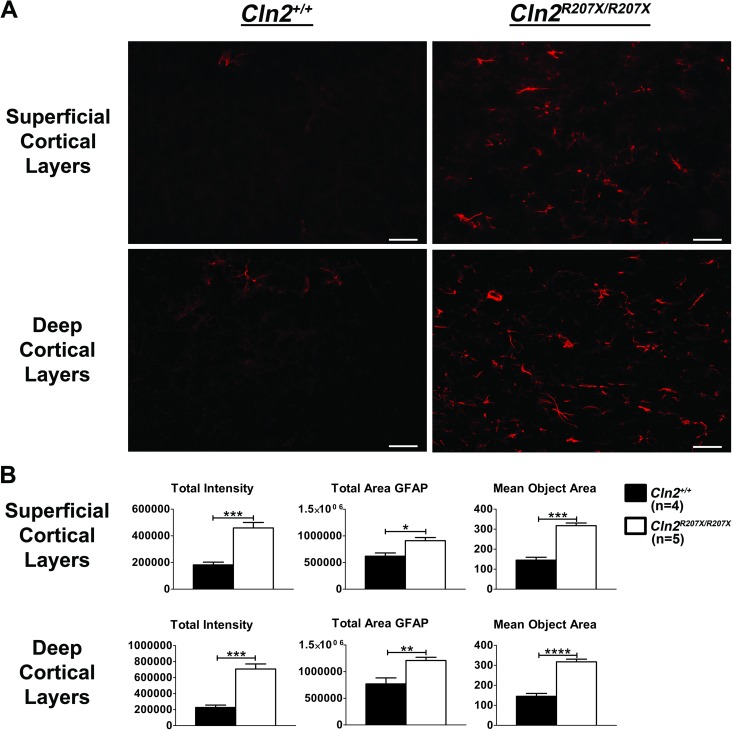
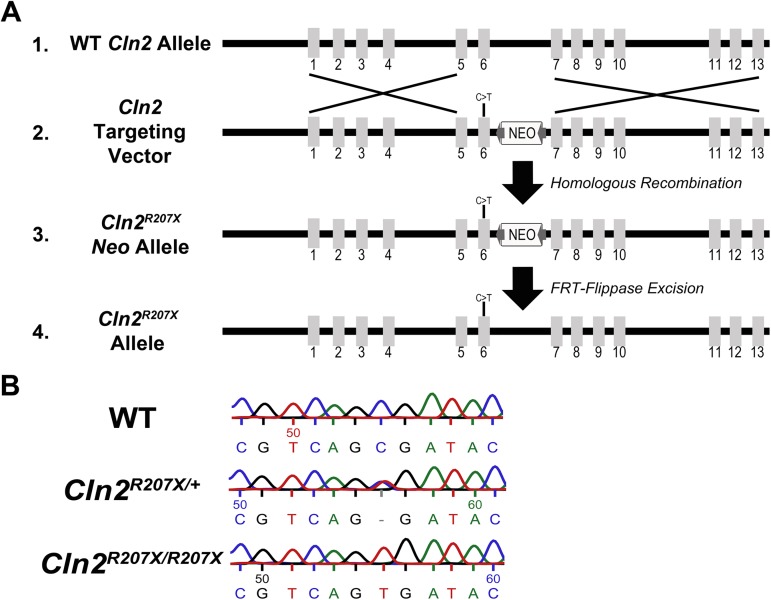
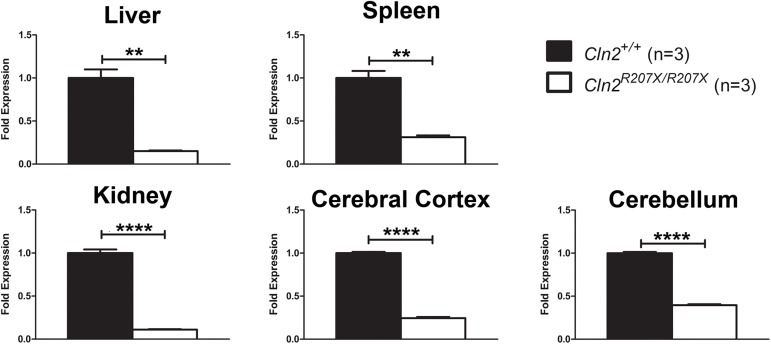
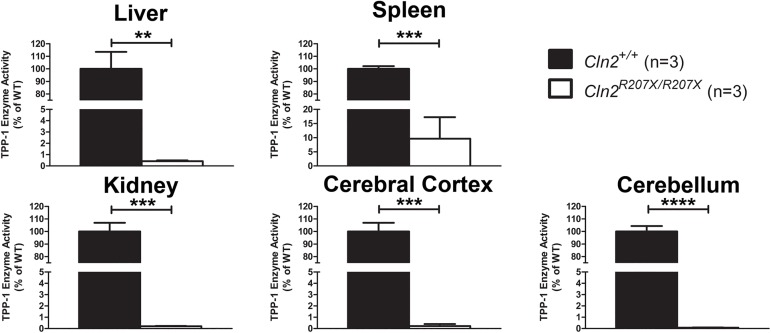
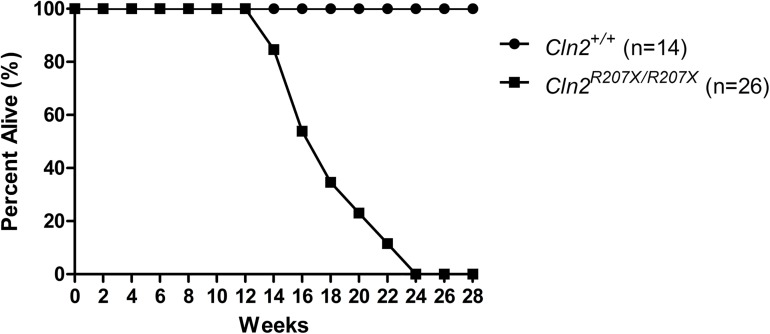
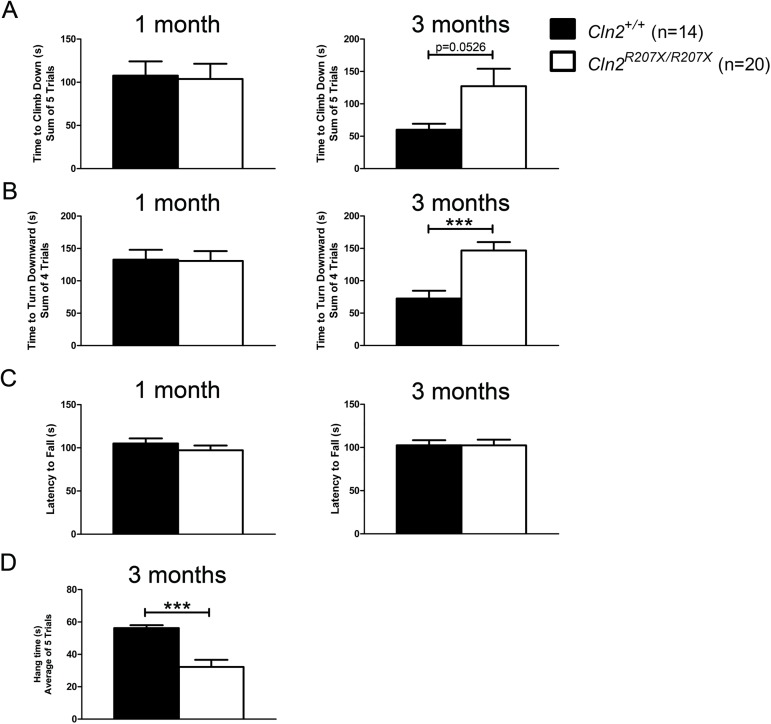
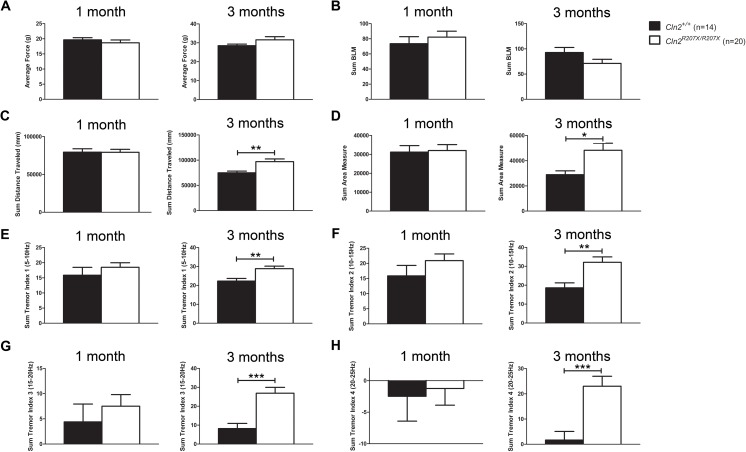
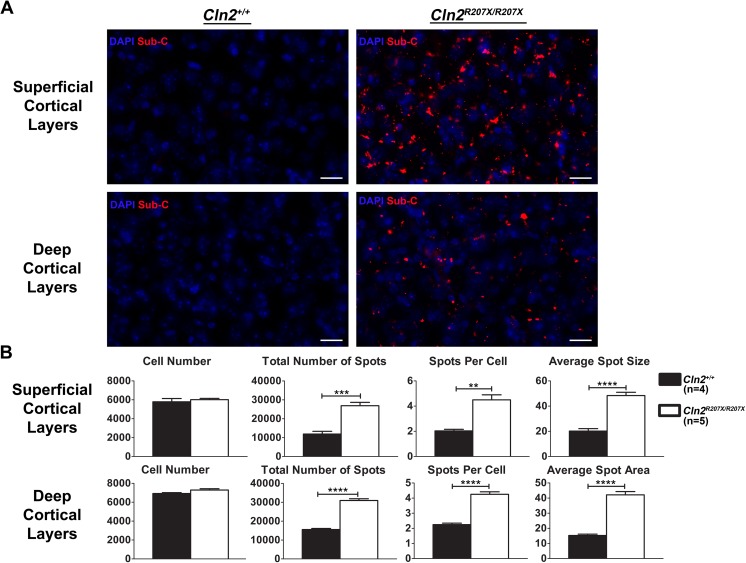
The Neuronal Ceroid Lipofuscinoses (NCLs), also known as Batten disease, result from mutations in over a dozen genes. Although, adults are susceptible, the NCLs are frequently classified as pediatric neurodegenerative diseases due to their greater pediatric prevalence. Initial clinical presentation usually consists of either seizures or retinopathy but develops to encompass both in conjunction with declining motor and cognitive function. The NCLs result in premature death due to the absence of curative therapies. Nevertheless, preclinical and clinical trials exist for various therapies. However, the genotypes of NCL animal models determine which therapeutic approaches can be assessed. Mutations of the CLN2 gene encoding a soluble lysosomal enzyme, tripeptidyl peptidase 1 (TPP1), cause late infantile NCL/CLN2 disease. The genotype of the original mouse model of CLN2 disease, Cln2-/-, excludes mutation guided therapies like antisense oligonucleotides and nonsense suppression. Therefore, the purpose of this study was to develop a model of CLN2 disease that allows for the assessment of all therapeutic approaches. Nonsense mutations in CLN2 disease are frequent, the most common being CLN2R208X. Thus, we created a mouse model that carries a mutation equivalent to the human p.R208X mutation. Molecular assessment of Cln2R207X/R207X tissues determined significant reduction in Cln2 transcript abundance and TPP1 enzyme activity. This reduction leads to the development of neurological impairment (e.g. tremors) and neuropathology (e.g. astrocytosis). Collectively, these assessments indicate that the Cln2R207X/R207X mouse is a valid CLN2 disease model which can be used for the preclinical evaluation of all therapeutic approaches including mutation guided therapies.
神经元蜡样脂褐质沉积症(NCLs),也被称为巴顿病,是由十几个基因的突变引起的。虽然成年人也易患此病,但由于其在儿童中的患病率更高,NCLs通常被归类为儿童神经退行性疾病。最初的临床表现通常包括癫痫发作或视网膜病变,但随后会发展为两者兼有,并伴有运动和认知功能下降。由于缺乏治愈性疗法,NCLs会导致过早死亡。尽管如此,针对各种疗法的临床前和临床试验都存在。然而,NCL动物模型的基因型决定了哪些治疗方法可以被评估。编码可溶性溶酶体酶三肽基肽酶1(TPP1)的CLN2基因突变会导致晚发性婴儿NCL/CLN2病。CLN2病的原始小鼠模型Cln2-/-的基因型排除了反义寡核苷酸和无义抑制等突变导向疗法。因此,本研究的目的是开发一种CLN2病模型,以评估所有治疗方法。CLN2病中的无义突变很常见,最常见的是CLN2R208X。因此,我们创建了一个携带与人类p.R208X突变等效突变的小鼠模型。对Cln2R207X/R207X组织的分子评估确定Cln转录本丰度和TPP1酶活性显著降低。这种降低导致神经功能障碍(如震颤)和神经病理学(如星形细胞增多)的发展。总体而言,这些评估表明Cln2R207X/R207X小鼠是一种有效的CLN2病模型,可用于包括突变导向疗法在内的所有治疗方法的临床前评估。