Department of Biomedical Informatics, Harvard Medical School, Boston, Massachusetts 02115, USA.
Botanical Institute, Biocenter, University of Cologne, D-50674 Cologne, Germany.
Nat Commun. 2017 May 5;8:15309. doi: 10.1038/ncomms15309.
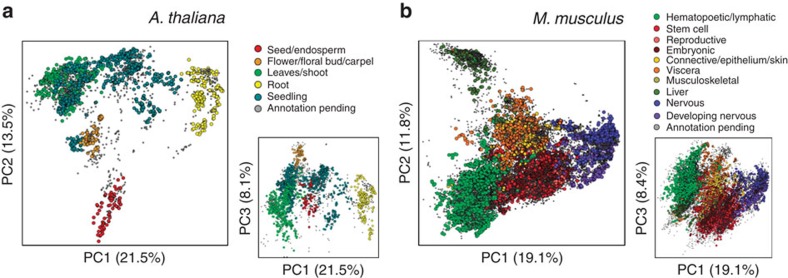
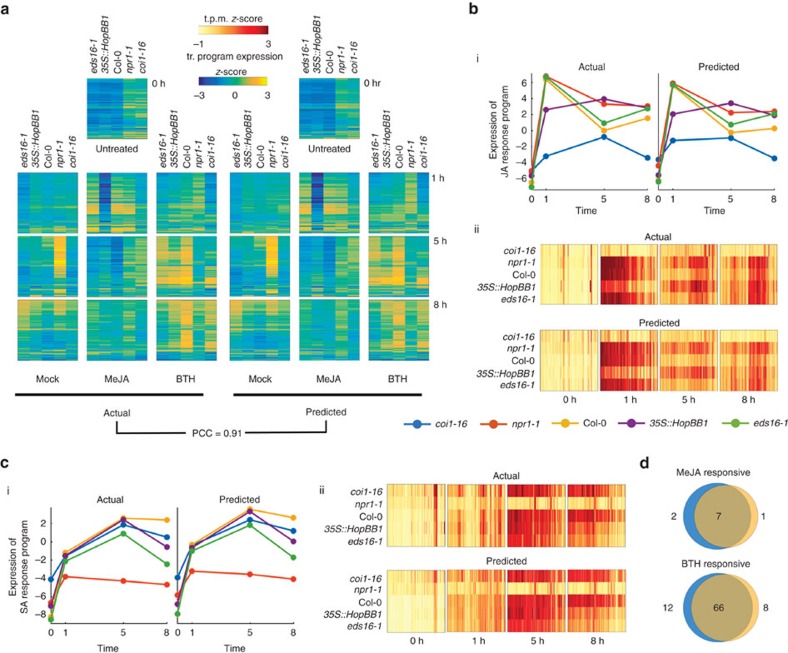
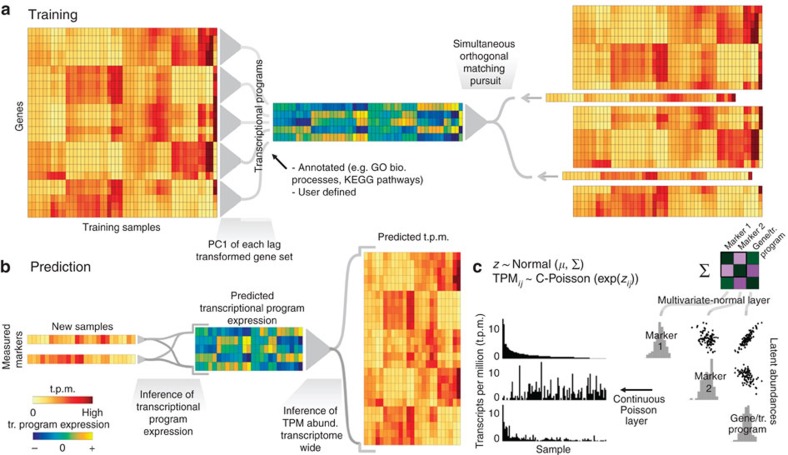
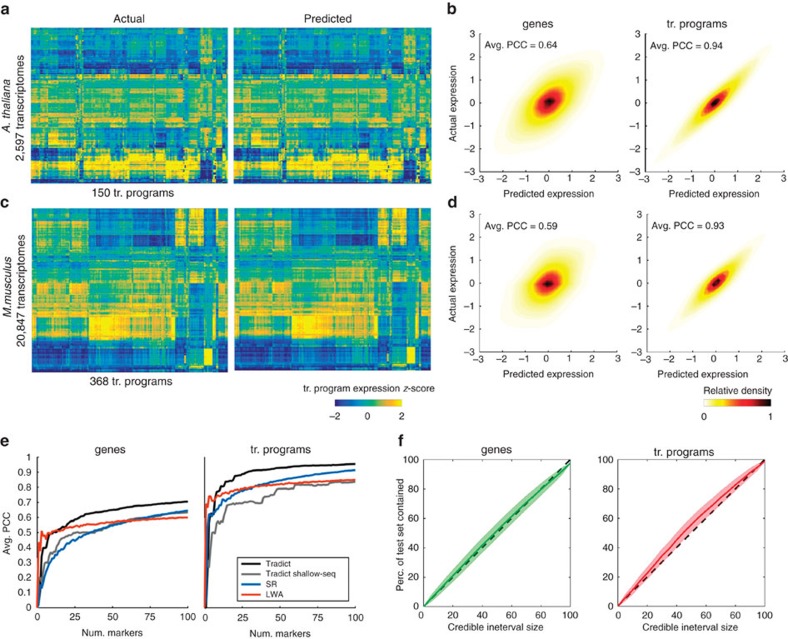
Transcript levels are a critical determinant of the proteome and hence cellular function. Because the transcriptome is an outcome of the interactions between genes and their products, it may be accurately represented by a subset of transcript abundances. We develop a method, Tradict (transcriptome predict), capable of learning and using the expression measurements of a small subset of 100 marker genes to predict transcriptome-wide gene abundances and the expression of a comprehensive, but interpretable list of transcriptional programs that represent the major biological processes and pathways of the cell. By analyzing over 23,000 publicly available RNA-Seq data sets, we show that Tradict is robust to noise and accurate. Coupled with targeted RNA sequencing, Tradict may therefore enable simultaneous transcriptome-wide screening and mechanistic investigation at large scales.
转录本水平是蛋白质组和细胞功能的关键决定因素。由于转录组是基因及其产物相互作用的结果,因此它可以通过转录本丰度的一个子集来准确表示。我们开发了一种方法 Tradict(转录组预测),能够学习和使用一小部分 100 个标记基因的表达测量值来预测全转录组基因丰度以及代表细胞主要生物学过程和途径的综合但可解释的转录程序的表达。通过分析超过 23000 个公开的 RNA-Seq 数据集,我们表明 Tradict 对噪声具有鲁棒性且准确。Tradict 与靶向 RNA 测序相结合,因此可能能够大规模同时进行全转录组筛选和机制研究。