Zimmermann Michael T, Kennedy Richard B, Grill Diane E, Oberg Ann L, Goergen Krista M, Ovsyannikova Inna G, Haralambieva Iana H, Poland Gregory A
Department of Health Science Research, Division of Biomedical Statistics and Informatics, Mayo Clinic, Rochester, MN, USA.
Mayo Clinic Vaccine Research Group, Mayo Clinic, Rochester, MN, USA.
Front Immunol. 2017 Apr 21;8:445. doi: 10.3389/fimmu.2017.00445. eCollection 2017.
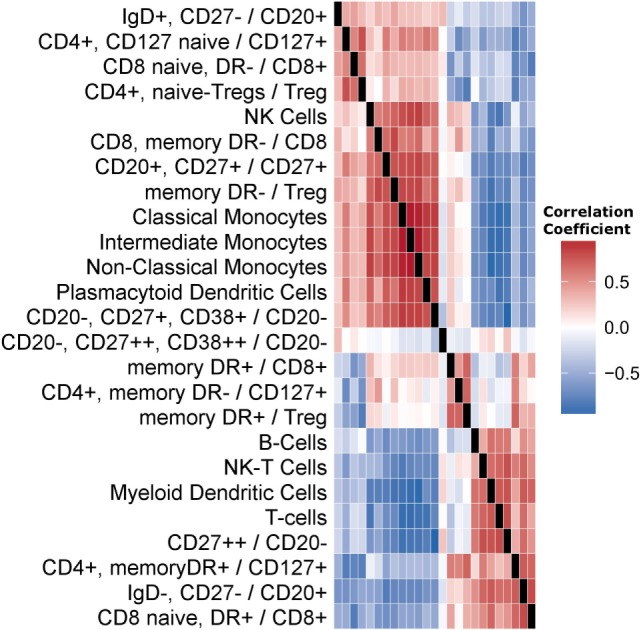
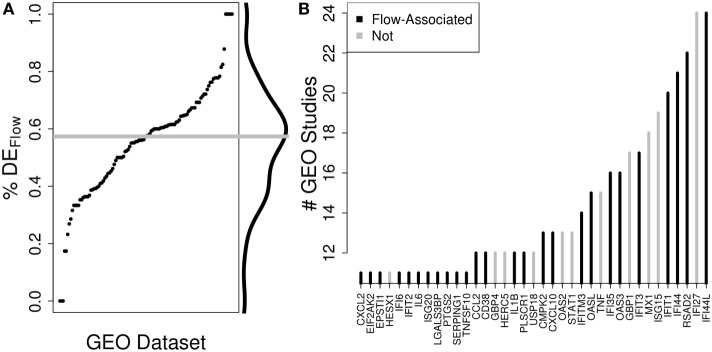
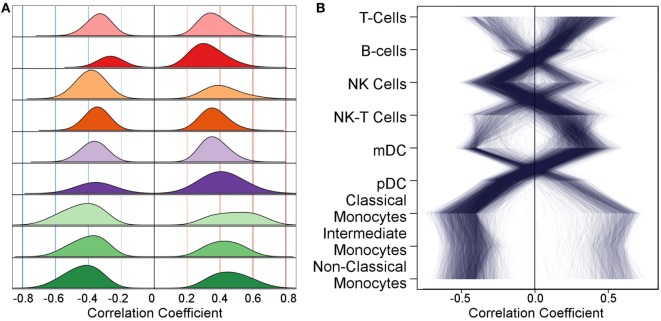
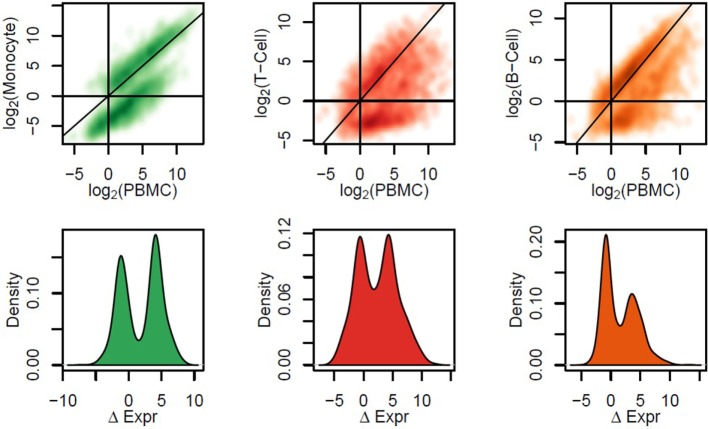
The development of a humoral immune response to influenza vaccines occurs on a multisystems level. Due to the orchestration required for robust immune responses when multiple genes and their regulatory components across multiple cell types are involved, we examined an influenza vaccination cohort using multiple high-throughput technologies. In this study, we sought a more thorough understanding of how immune cell composition and gene expression relate to each other and contribute to interindividual variation in response to influenza vaccination. We first hypothesized that many of the differentially expressed (DE) genes observed after influenza vaccination result from changes in the composition of participants' peripheral blood mononuclear cells (PBMCs), which were assessed using flow cytometry. We demonstrated that DE genes in our study are correlated with changes in PBMC composition. We gathered DE genes from 128 other publically available PBMC-based vaccine studies and identified that an average of 57% correlated with specific cell subset levels in our study (permutation used to control false discovery), suggesting that the associations we have identified are likely general features of PBMC-based transcriptomics. Second, we hypothesized that more robust models of vaccine response could be generated by accounting for the interplay between PBMC composition, gene expression, and gene regulation. We employed machine learning to generate predictive models of B-cell ELISPOT response outcomes and hemagglutination inhibition (HAI) antibody titers. The top HAI and B-cell ELISPOT model achieved an area under the receiver operating curve (AUC) of 0.64 and 0.79, respectively, with linear model coefficients of determination of 0.08 and 0.28. For the B-cell ELISPOT outcomes, CpG methylation had the greatest predictive ability, highlighting potentially novel regulatory features important for immune response. B-cell ELISOT models using only PBMC composition had lower performance (AUC = 0.67), but highlighted well-known mechanisms. Our analysis demonstrated that each of the three data sets (cell composition, mRNA-Seq, and DNA methylation) may provide distinct information for the prediction of humoral immune response outcomes. We believe that these findings are important for the interpretation of current omics-based studies and set the stage for a more thorough understanding of interindividual immune responses to influenza vaccination.
对流感疫苗的体液免疫反应在多系统水平上发生。由于当涉及多种细胞类型中的多个基因及其调控成分时,强大的免疫反应需要精心协调,我们使用多种高通量技术对一个流感疫苗接种队列进行了研究。在本研究中,我们试图更全面地了解免疫细胞组成和基因表达如何相互关联,并导致个体对流感疫苗反应的差异。我们首先假设,流感疫苗接种后观察到的许多差异表达(DE)基因是由于参与者外周血单个核细胞(PBMC)组成的变化所致,我们使用流式细胞术对其进行了评估。我们证明,我们研究中的DE基因与PBMC组成的变化相关。我们从其他128项基于PBMC的公开可用疫苗研究中收集了DE基因,并确定平均57%与我们研究中的特定细胞亚群水平相关(使用置换法控制错误发现率),这表明我们所确定的关联可能是基于PBMC的转录组学的普遍特征。其次,我们假设通过考虑PBMC组成、基因表达和基因调控之间的相互作用,可以生成更强大的疫苗反应模型。我们使用机器学习生成B细胞ELISPOT反应结果和血凝抑制(HAI)抗体滴度的预测模型。最佳的HAI和B细胞ELISPOT模型的受试者操作特征曲线下面积(AUC)分别为0.64和0.79,线性模型的决定系数分别为0.08和0.28。对于B细胞ELISPOT结果,CpG甲基化具有最大的预测能力,突出了对免疫反应重要的潜在新调控特征。仅使用PBMC组成的B细胞ELISOT模型性能较低(AUC = 0.67),但突出了已知机制。我们的分析表明,三个数据集(细胞组成、mRNA测序和DNA甲基化)中的每一个都可能为预测体液免疫反应结果提供独特的信息。我们认为,这些发现对于解释当前基于组学的研究很重要,并为更全面地了解个体对流感疫苗的免疫反应奠定了基础。