Dolgalev Igor, Sedlazeck Fritz, Busby Ben
New York University School of Medicine, New York, NY, 10016, USA.
Department of Computer Science, Johns Hopkins University, Baltimore, MD, 21202, USA.
F1000Res. 2017 Apr 7;6:443. doi: 10.12688/f1000research.11254.1. eCollection 2017.
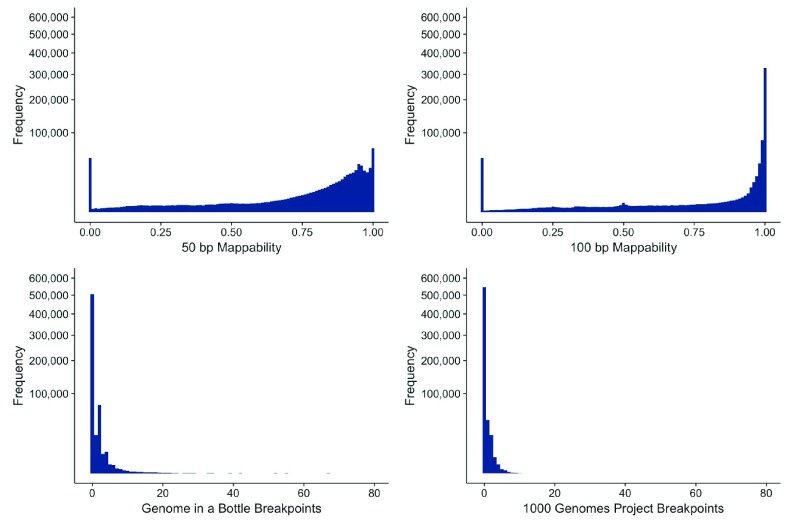
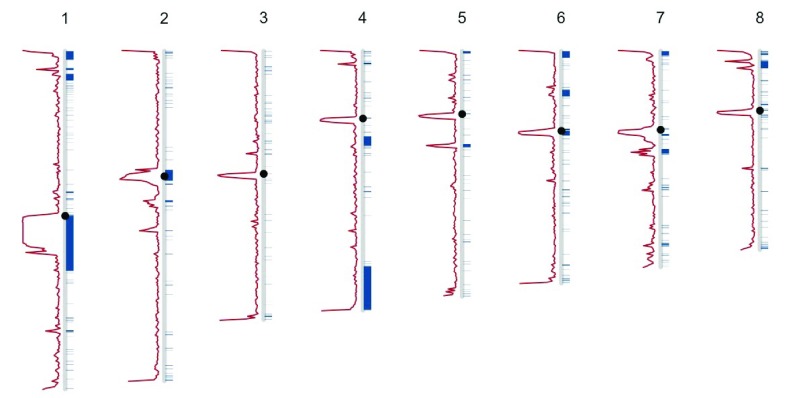
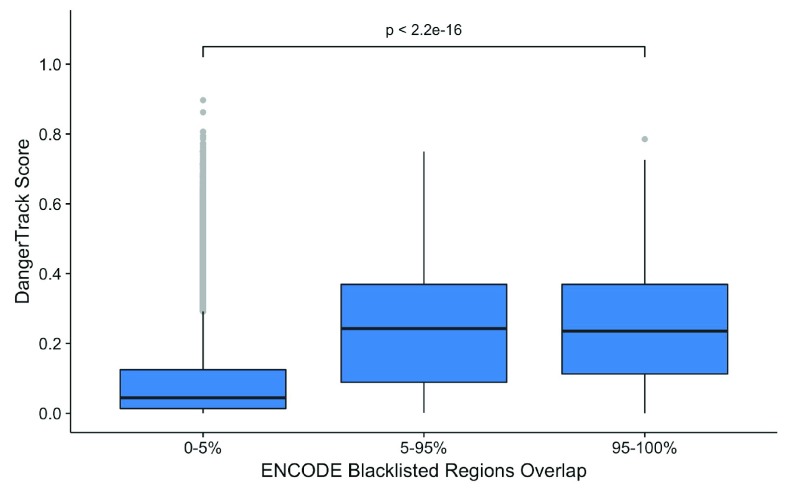
Over recent years, multiple groups have shown that a large number of structural variants, repeats, or problems with the underlying genome assembly have dramatic effects on the mapping, calling, and overall reliability of single nucleotide polymorphism calls. This project endeavored to develop an easy-to-use track for looking at structural variant and repeat regions. This track, DangerTrack, can be displayed alongside the existing Genome Reference Consortium assembly tracks to warn clinicians and biologists when variants of interest may be incorrectly called, of dubious quality, or on an insertion or copy number expansion. While mapping and variant calling can be automated, it is our opinion that when these regions are of interest to a particular clinical or research group, they warrant a careful examination, potentially involving localized reassembly. DangerTrack is available at https://github.com/DCGenomics/DangerTrack.
近年来,多个研究团队表明,大量的结构变异、重复序列或基础基因组组装问题对单核苷酸多态性位点的定位、识别以及整体可靠性有着显著影响。该项目致力于开发一个易于使用的轨迹,用于查看结构变异和重复区域。这个名为“危险轨迹(DangerTrack)”的轨迹可以与现有的基因组参考联盟组装轨迹一起显示,以便在感兴趣的变异可能被错误识别、质量存疑,或者存在插入或拷贝数扩增时,向临床医生和生物学家发出警告。虽然定位和变异识别可以自动化,但我们认为,当这些区域对特定的临床或研究团队有意义时,它们需要仔细检查,可能还需要进行局部重新组装。可通过https://github.com/DCGenomics/DangerTrack获取“危险轨迹”。