Alhakami Hind, Mirebrahim Hamid, Lonardi Stefano
Department of Computer Science & Engineering, University of California, 900 University Avenue, Riverside, 92521, CA, USA.
Genome Biol. 2017 May 18;18(1):93. doi: 10.1186/s13059-017-1213-3.
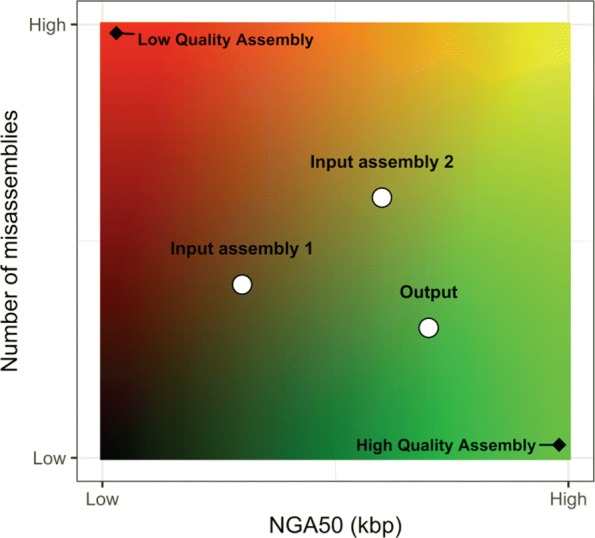
The majority of eukaryotic genomes are unfinished due to the algorithmic challenges of assembling them. A variety of assembly and scaffolding tools are available, but it is not always obvious which tool or parameters to use for a specific genome size and complexity. It is, therefore, common practice to produce multiple assemblies using different assemblers and parameters, then select the best one for public release. A more compelling approach would allow one to merge multiple assemblies with the intent of producing a higher quality consensus assembly, which is the objective of assembly reconciliation.
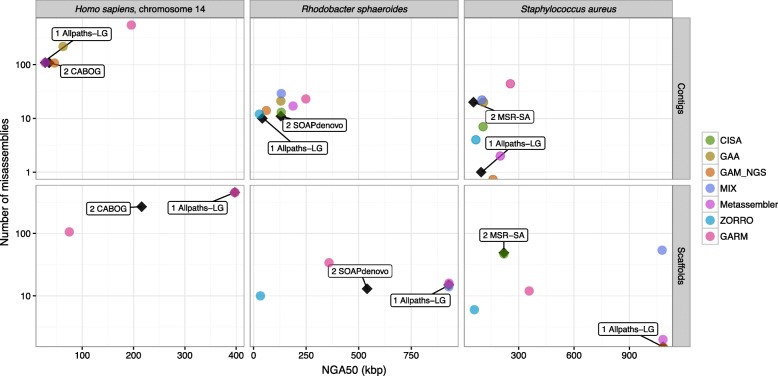
Several assembly reconciliation tools have been proposed in the literature, but their strengths and weaknesses have never been compared on a common dataset. We fill this need with this work, in which we report on an extensive comparative evaluation of several tools. Specifically, we evaluate contiguity, correctness, coverage, and the duplication ratio of the merged assembly compared to the individual assemblies provided as input.
None of the tools we tested consistently improved the quality of the input GAGE and synthetic assemblies. Our experiments show an increase in contiguity in the consensus assembly when the original assemblies already have high quality. In terms of correctness, the quality of the results depends on the specific tool, as well as on the quality and the ranking of the input assemblies. In general, the number of misassemblies ranges from being comparable to the best of the input assembly to being comparable to the worst of the input assembly.
由于组装算法上的挑战,大多数真核生物基因组尚未完成。有多种组装和支架搭建工具可供使用,但对于特定的基因组大小和复杂度,使用哪种工具或参数并不总是显而易见的。因此,常见的做法是使用不同的组装器和参数生成多个组装结果,然后选择最佳的一个用于公开发布。一种更具吸引力的方法是将多个组装结果合并,以生成更高质量的一致性组装结果,这就是组装结果协调的目标。
文献中已经提出了几种组装结果协调工具,但它们的优缺点从未在一个通用数据集上进行过比较。我们通过这项工作满足了这一需求,在其中我们报告了对几种工具的广泛比较评估。具体而言,我们评估了合并后的组装结果与作为输入提供的各个组装结果相比的连续性、正确性、覆盖率和重复率。
我们测试的工具中没有一个能始终如一地提高输入的GAGE和合成组装结果的质量。我们的实验表明,当原始组装结果已经具有高质量时,一致性组装结果的连续性会增加。在正确性方面,结果的质量取决于特定的工具,以及输入组装结果的质量和排名。一般来说,错误组装的数量范围从与输入组装结果中最好的相当到与输入组装结果中最差的相当。