Strongly Correlated Systems "Lendület" Research Group, Institute for Solid State Physics and Optics, MTA Wigner Research Centre for Physics, H-1121, Budapest, Konkoly-Thege Miklós út 29-33, Hungary.
Department of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, Wisconsin, 53706, United States.
Sci Rep. 2017 May 22;7(1):2237. doi: 10.1038/s41598-017-02447-z.
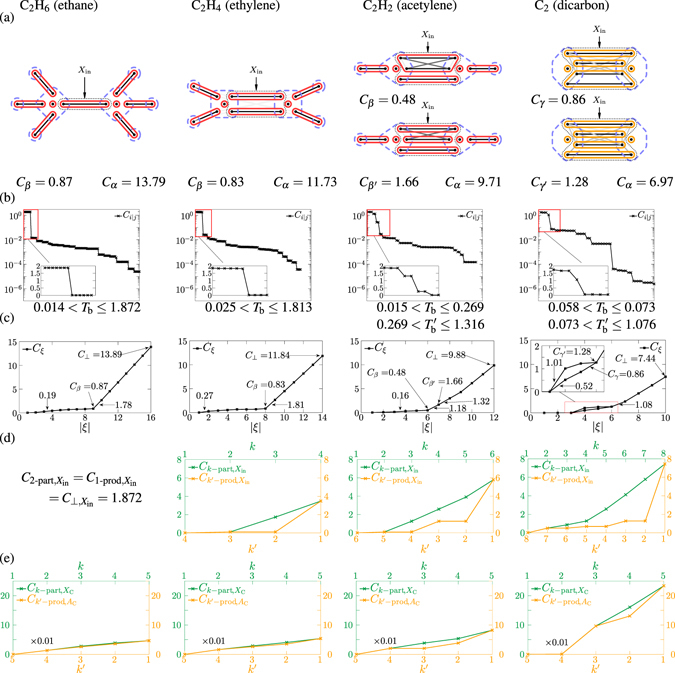
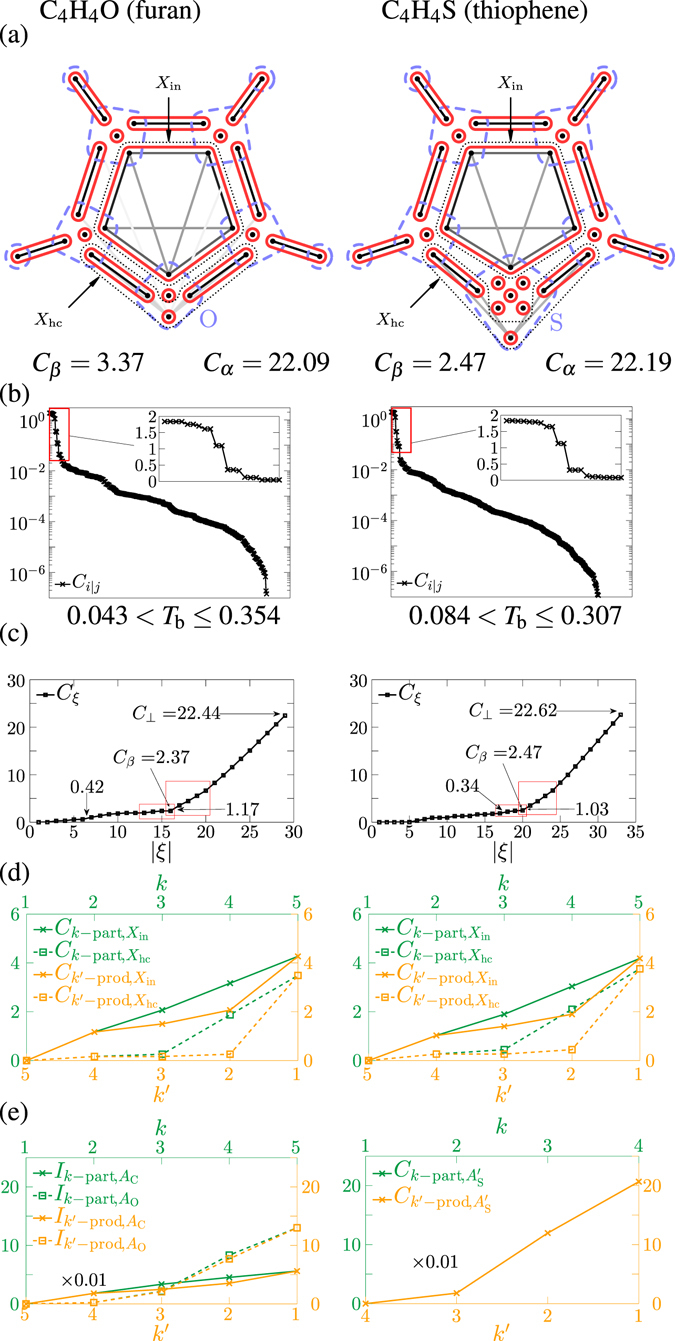
The quantum mechanical description of the chemical bond is generally given in terms of delocalized bonding orbitals, or, alternatively, in terms of correlations of occupations of localised orbitals. However, in the latter case, multiorbital correlations were treated only in terms of two-orbital correlations, although the structure of multiorbital correlations is far richer; and, in the case of bonds established by more than two electrons, multiorbital correlations represent a more natural point of view. Here, for the first time, we introduce the true multiorbital correlation theory, consisting of a framework for handling the structure of multiorbital correlations, a toolbox of true multiorbital correlation measures, and the formulation of the multiorbital correlation clustering, together with an algorithm for obtaining that. These make it possible to characterise quantitatively, how well a bonding picture describes the chemical system. As proof of concept, we apply the theory for the investigation of the bond structures of several molecules. We show that the non-existence of well-defined multiorbital correlation clustering provides a reason for debated bonding picture.
化学键的量子力学描述通常是用离域成键轨道来表示的,或者用局部轨道占据的相关性来表示。然而,在后一种情况下,虽然多轨道相关性的结构要丰富得多,但只在双轨道相关性的基础上处理多轨道相关性;并且,对于由两个以上电子建立的键,多轨道相关性代表了一个更自然的观点。在这里,我们首次引入了真正的多轨道相关理论,它由一个用于处理多轨道相关性结构的框架、一套真正的多轨道相关度量工具以及多轨道相关聚类的表述以及获得该聚类的算法组成。这些使得可以定量地描述成键图像对化学系统的描述程度。作为概念验证,我们将该理论应用于几个分子的键结构研究。我们表明,不存在明确定义的多轨道相关聚类为有争议的成键图像提供了一个理由。