Hylleraas Centre for Quantum Molecular Sciences, Department of Chemistry , University of Oslo , P.O. Box 1033 Blindern, N-0315 Oslo , Norway.
Strongly Correlated Systems "Lendület" Research Group , Wigner Research Center for Physics , H-1525 , P.O. Box 49, Budapest , Hungary.
J Chem Theory Comput. 2019 Apr 9;15(4):2206-2220. doi: 10.1021/acs.jctc.8b00960. Epub 2019 Mar 13.
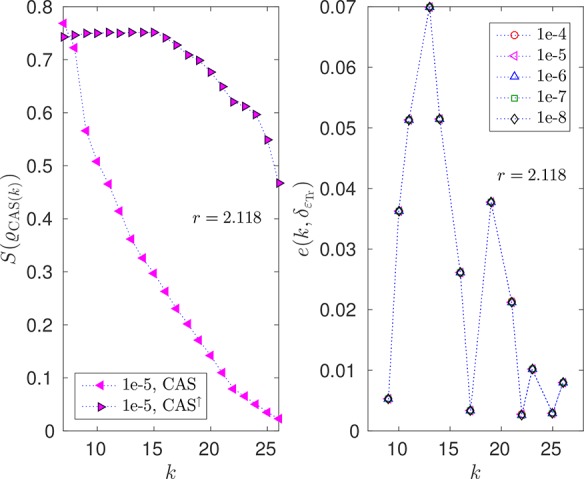
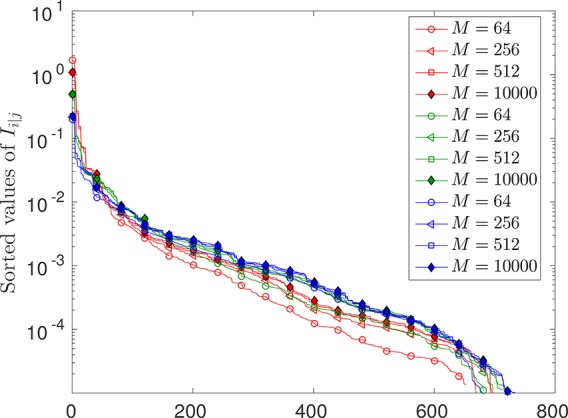
In this article, we investigate the numerical and theoretical aspects of the coupled-cluster method tailored by matrix-product states. We investigate formal properties of the used method, such as energy size consistency and the equivalence of linked and unlinked formulation. The existing mathematical analysis is here elaborated in a quantum chemical framework. In particular, we highlight the use of what we have defined as a complete active space-external space gap describing the basis splitting between the complete active space and the external part generalizing the concept of a HOMO-LUMO gap. Furthermore, the behavior of the energy error for an optimal basis splitting, i.e., an active space choice minimizing the density matrix renormalization group-tailored coupled-cluster singles doubles error, is discussed. We show numerical investigations on the robustness with respect to the bond dimensions of the single orbital entropy and the mutual information, which are quantities that are used to choose a complete active space. Moreover, the dependence of the ground-state energy error on the complete active space has been analyzed numerically in order to find an optimal split between the complete active space and external space by minimizing the density matrix renormalization group-tailored coupled-cluster error.
在本文中,我们研究了通过矩阵乘积态定制的耦合簇方法的数值和理论方面。我们研究了所使用方法的形式性质,例如能量大小一致性和链接与非链接公式的等价性。现有的数学分析在量子化学框架中得到了阐述。特别是,我们强调了使用我们定义的完全活性空间-外部空间间隙来描述完全活性空间和外部部分之间的基分裂,该间隙推广了 HOMO-LUMO 间隙的概念。此外,还讨论了最优基分裂(即通过密度矩阵重整化群定制的耦合簇单双误差最小化来选择的活性空间)的能量误差的行为。我们对单轨道熵和互信息的稳健性进行了数值研究,这两个量被用来选择完全活性空间。此外,还分析了基态能量误差对完全活性空间的依赖性,以便通过最小化密度矩阵重整化群定制的耦合簇误差来找到完全活性空间和外部空间之间的最佳分裂。