Konigorski Stefan, Yilmaz Yildiz E, Pischon Tobias
Molecular Epidemiology Research Group, Max Delbrück Center (MDC) for Molecular Medicine in the Helmholtz Association, Berlin, Germany.
Department of Mathematics and Statistics, Memorial University of Newfoundland, St. John's, Newfoundland and Labrador, Canada.
PLoS One. 2017 May 31;12(5):e0178504. doi: 10.1371/journal.pone.0178504. eCollection 2017.
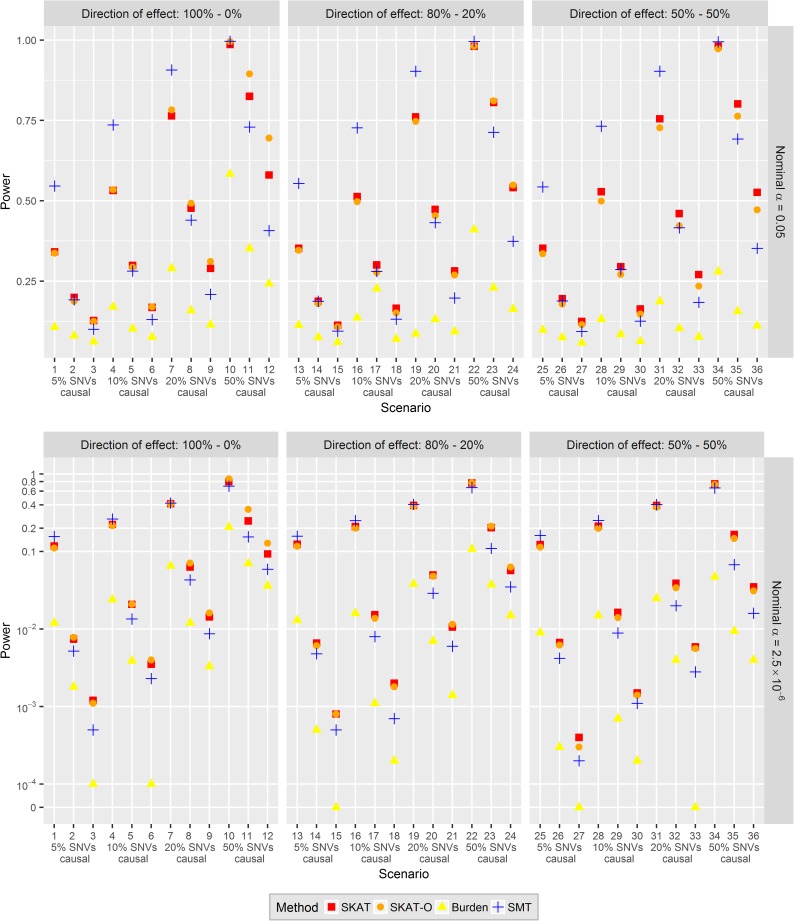
In genetic association studies of rare variants, low statistical power and potential violations of established estimator properties are among the main challenges of association tests. Multi-marker tests (MMTs) have been proposed to target these challenges, but any comparison with single-marker tests (SMTs) has to consider that their aim is to identify causal genomic regions instead of variants. Valid power comparisons have been performed for the analysis of binary traits indicating that MMTs have higher power, but there is a lack of conclusive studies for quantitative traits. The aim of our study was therefore to fairly compare SMTs and MMTs in their empirical power to identify the same causal loci associated with a quantitative trait. The results of extensive simulation studies indicate that previous results for binary traits cannot be generalized. First, we show that for the analysis of quantitative traits, conventional estimation methods and test statistics of single-marker approaches have valid properties yielding association tests with valid type I error, even when investigating singletons or doubletons. Furthermore, SMTs lead to more powerful association tests for identifying causal genes than MMTs when the effect sizes of causal variants are large, and less powerful tests when causal variants have small effect sizes. For moderate effect sizes, whether SMTs or MMTs have higher power depends on the sample size and percentage of causal SNVs. For a more complete picture, we also compare the power in studies of quantitative and binary traits, and the power to identify causal genes with the power to identify causal rare variants. In a genetic association analysis of systolic blood pressure in the Genetic Analysis Workshop 19 data, SMTs yielded smaller p-values compared to MMTs for most of the investigated blood pressure genes, and were least influenced by the definition of gene regions.
在罕见变异的基因关联研究中,统计功效低以及既定估计量属性可能被违反是关联检验的主要挑战。为应对这些挑战,人们提出了多标记检验(MMT),但与单标记检验(SMT)进行任何比较时都必须考虑到,MMT的目的是识别因果基因组区域而非变异。对于二元性状分析已经进行了有效的功效比较,结果表明MMT具有更高的功效,但对于定量性状缺乏结论性研究。因此,我们研究的目的是公平比较SMT和MMT在识别与定量性状相关的相同因果位点方面的实证功效。广泛的模拟研究结果表明,之前关于二元性状的结果不能一概而论。首先,我们表明,对于定量性状分析,单标记方法的传统估计方法和检验统计量具有有效的属性,即使在研究单倍体或双体时也能产生具有有效I型错误的关联检验。此外,当因果变异的效应大时,SMT在识别因果基因方面比MMT能产生更有效的关联检验;当因果变异的效应小时,SMT的检验功效则较低。对于中等效应大小,SMT还是MMT具有更高的功效取决于样本量和因果单核苷酸变异(SNV)的百分比。为了更全面地了解情况,我们还比较了定量和二元性状研究中的功效,以及识别因果基因的功效与识别因果罕见变异的功效。在遗传分析研讨会19数据中对收缩压进行的基因关联分析中,对于大多数研究的血压基因,与MMT相比,SMT产生的p值更小,并且受基因区域定义的影响最小。