Matsuda Fumio, Tomita Atsumi, Shimizu Hiroshi
Department of Bioinformatic Engineering, Graduate School of Information Science and Technology, Osaka University.
RIKEN Center for Sustainable Resource Science.
Mass Spectrom (Tokyo). 2017;6(1):A0056. doi: 10.5702/massspectrometry.A0056. Epub 2017 Jun 2.
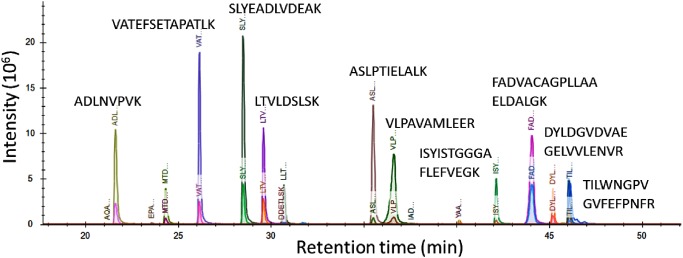
In targeted proteomics using liquid chromatography-tandem triple quadrupole mass spectrometry (LC/MS/MS) in the selected reaction monitoring (SRM) mode, selecting the best observable or visible peptides is a key step in the development of SRM assay methods of target proteins. A direct comparison of signal intensities among all candidate peptides by brute-force LC/MS/MS analysis is a concrete approach for peptide selection. However, the analysis requires an SRM method with hundreds of transitions. This study reports on the development of a method for predicting and identifying hopeless peptides to reduce the number of candidate peptides needed for brute-force experiments. Hopeless peptides are proteotypic peptides that are unlikely to be selected for targets in SRM analysis owing to their poor ionization characteristics. Targeted proteomics data from demonstrated that the relative ionization efficiency between two peptides could be predicted from sequences of two peptides, when a multivariate regression model is used. Validation of the method showed that >20% of the candidate peptides could be successfully eliminated as hopeless peptides with a false positive rate of less than 2%.
在采用液相色谱-串联三重四极杆质谱(LC/MS/MS)的选择反应监测(SRM)模式进行靶向蛋白质组学研究时,选择最佳可观测或可见肽段是目标蛋白质SRM分析方法开发中的关键步骤。通过强力LC/MS/MS分析直接比较所有候选肽段之间的信号强度是肽段选择的具体方法。然而,该分析需要具有数百个跃迁的SRM方法。本研究报告了一种预测和识别无望肽段的方法的开发,以减少强力实验所需的候选肽段数量。无望肽段是由于其电离特性较差而不太可能在SRM分析中被选作目标的蛋白型肽段。来自 的靶向蛋白质组学数据表明,当使用多元回归模型时,可以从两个肽段的序列预测两个肽段之间的相对电离效率。该方法的验证表明,超过20%的候选肽段可作为无望肽段成功消除,假阳性率低于2%。