Flevaris Panagiotis, Khan Sadiya S, Eren Mesut, Schuldt Adam J T, Shah Sanjiv J, Lee Daniel C, Gupta Sweta, Shapiro Amy D, Burridge Paul W, Ghosh Asish K, Vaughan Douglas E
From Division of Cardiology, Department of Medicine (P.F., S.S.K., M.E., A.J.T.S., S.J.S., D.C.L., A.K.G., D.E.V.); Feinberg Cardiovascular Research Institute (P.F., S.S.K., S.J.S., D.C.L., A.K.G., D.E.V.), Department of Pharmacology (A.J.T.S., P.W.B.), Northwestern University Feinberg School of Medicine, Chicago, IL; and Indiana Hemophilia and Thrombosis Center, Indianapolis (S.G., A.D.S.).
Circulation. 2017 Aug 15;136(7):664-679. doi: 10.1161/CIRCULATIONAHA.117.028145. Epub 2017 Jun 6.
Fibrosis is the pathological consequence of stress-induced tissue remodeling and matrix accumulation. Increased levels of plasminogen activator inhibitor type I (PAI-1) have been shown to promote fibrosis in multiple organ systems. Paradoxically, homozygous genetic deficiency of PAI-1 is associated with spontaneous age-dependent, cardiac-selective fibrosis in mice. We have identified a novel PAI-1-dependent mechanism that regulates cardiomyocyte-derived fibrogenic signals and cardiac transcriptional pathways during injury.
Cardiac fibrosis in subjects with homozygous mutation in was evaluated with late gadolinium-enhanced cardiac magnetic resonance imaging. A murine cardiac injury model was performed by subcutaneous infusion of either saline or Angiotensin II by osmotic minipumps. We evaluated blood pressure, cardiac function (by echocardiography), fibrosis (with Masson Trichrome staining), and apoptosis (with TUNEL staining), and we performed transcriptome analysis (with RNA sequencing). We further evaluated fibrotic signaling in isolated murine primary ventricular myocytes.
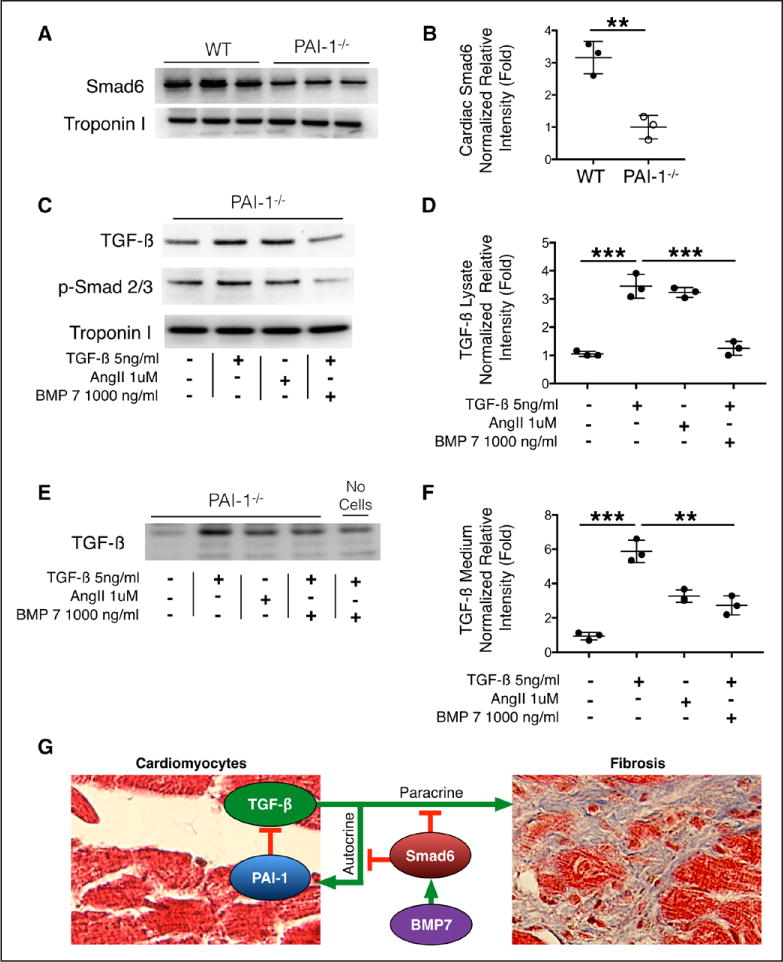
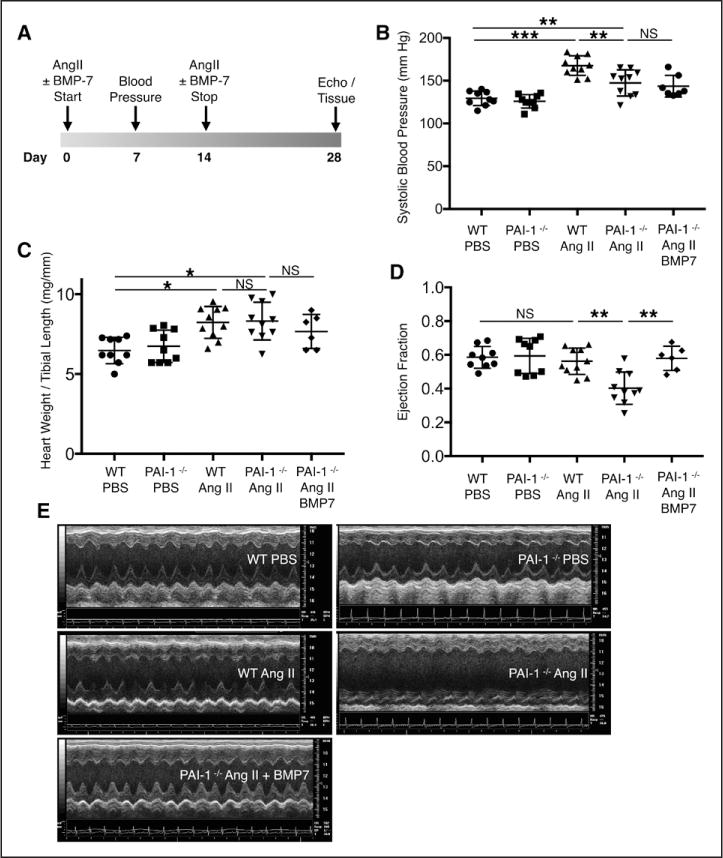
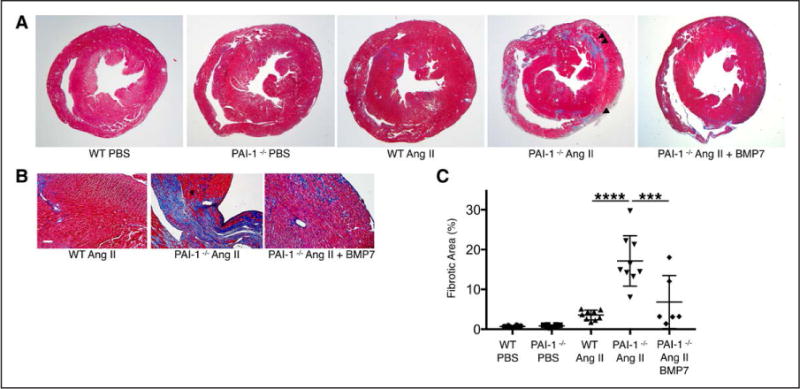
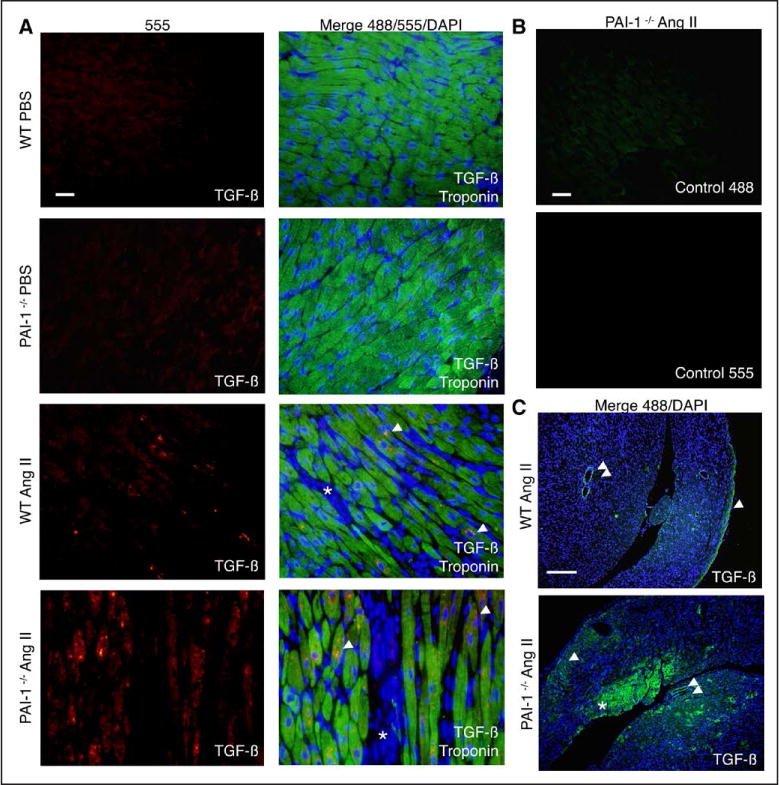
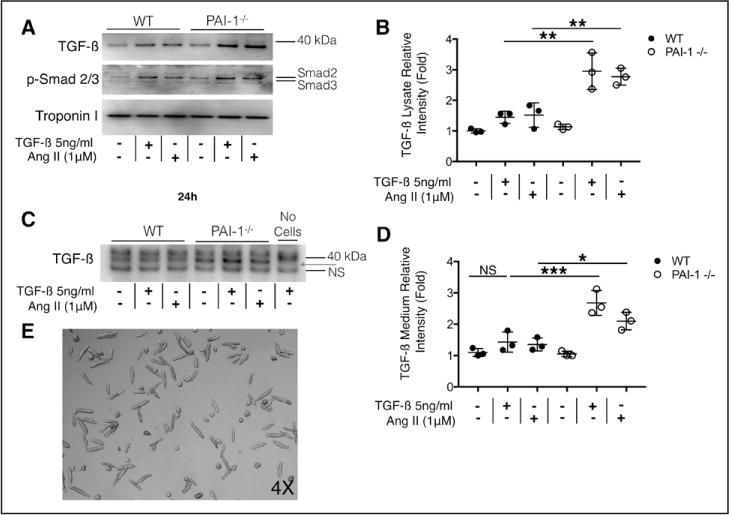
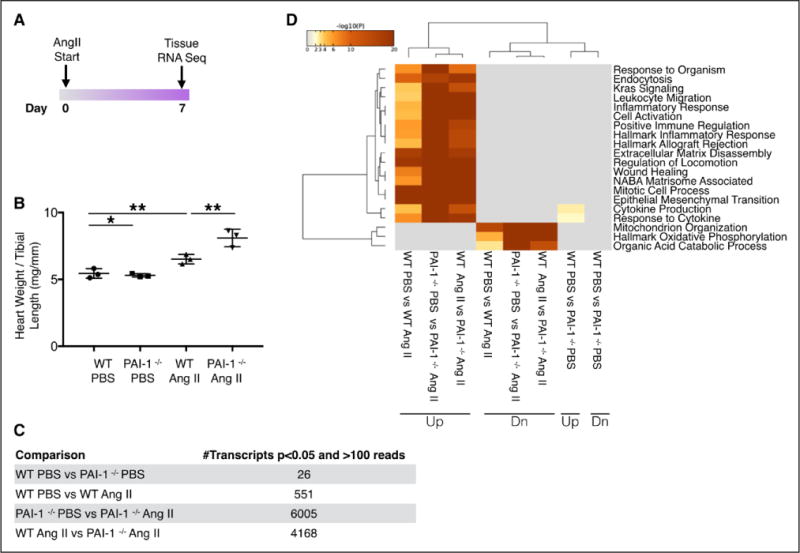
Cardiac fibrosis was detected in 2 otherwise healthy humans with complete PAI-1 deficiency because of a homozygous frameshift mutation in . In addition to its suppressive role during spontaneous cardiac fibrosis in multiple species, we hypothesized that PAI-1 also regulates fibrosis during cardiac injury. Treatment of young PAI-1 mice with Angiotensin II induced extensive hypertrophy and fibrotic cardiomyopathy, with increased cardiac apoptosis and both reactive and replacement fibrosis. Although Angiotensin II-induced hypertension was blunted in PAI-1 mice, cardiac hypertrophy was accelerated. Furthermore, ventricular myocytes were found to be an important source of cardiac transforming growth factor-β (TGF-β) and PAI-1 regulated TGF-β synthesis by cardiomyocytes in vitro as well as in vivo during cardiac injury. Transcriptome analysis of ventricular RNA after Angiotensin II treatment confirmed that PAI-1 deficiency significantly enhanced multiple TGF-β signaling elements and transcriptional targets, including genes for extracellular matrix components, mediators of extracellular matrix remodeling, matricellular proteins, and cardiac integrins compared with wild-type mice.
PAI-1 is an essential repressor of cardiac fibrosis in mammals. We define a novel cardiomyocyte-specific regulatory mechanism for TGF-β production by PAI-1, which explains the paradoxical effect of PAI-1 deficiency in promoting cardiac-selective fibrosis. Thus, PAI-1 is a molecular switch that controls the cardiac TGF-β axis and its early transcriptional effects that lead to myocardial fibrosis.
纤维化是应激诱导的组织重塑和基质积累的病理后果。已表明纤溶酶原激活物抑制剂I型(PAI-1)水平升高可促进多个器官系统的纤维化。矛盾的是,PAI-1纯合基因缺陷与小鼠自发性年龄依赖性心脏选择性纤维化有关。我们发现了一种新的PAI-1依赖性机制,该机制在损伤期间调节心肌细胞衍生的纤维化信号和心脏转录途径。
采用延迟钆增强心脏磁共振成像评估具有 纯合突变受试者的心脏纤维化。通过渗透微型泵皮下注射生理盐水或血管紧张素II建立小鼠心脏损伤模型。我们评估了血压、心脏功能(通过超声心动图)、纤维化(采用Masson三色染色)和细胞凋亡(采用TUNEL染色),并进行了转录组分析(采用RNA测序)。我们进一步评估了分离的小鼠原代心室肌细胞中的纤维化信号传导。
在2名因 纯合移码突变而完全缺乏PAI-1的健康人中检测到心脏纤维化。除了其在多个物种自发性心脏纤维化过程中的抑制作用外,我们推测PAI-1在心脏损伤期间也调节纤维化。用血管紧张素II处理年轻的PAI-1基因敲除小鼠会诱导广泛的肥大和纤维化性心肌病,心脏细胞凋亡增加,同时出现反应性纤维化和替代性纤维化。尽管血管紧张素II诱导的高血压在PAI-1基因敲除小鼠中减弱,但心脏肥大加速。此外,心室肌细胞被发现是心脏转化生长因子-β(TGF-β)的重要来源,并且PAI-1在体外以及心脏损伤期间的体内均调节心肌细胞的TGF-β合成。血管紧张素II处理后心室RNA的转录组分析证实,与野生型小鼠相比,PAI-1缺乏显著增强了多个TGF-β信号元件和转录靶点,包括细胞外基质成分、细胞外基质重塑介质、基质细胞蛋白和心脏整合素的基因。
PAI-1是哺乳动物心脏纤维化的重要抑制因子。我们定义了一种由PAI-1产生TGF-β的新的心肌细胞特异性调节机制,这解释了PAI-1缺乏在促进心脏选择性纤维化中的矛盾作用。因此,PAI-1是一个分子开关,控制心脏TGF-β轴及其导致心肌纤维化的早期转录效应。