Castro-Mondragon Jaime Abraham, Jaeger Sébastien, Thieffry Denis, Thomas-Chollier Morgane, van Helden Jacques
Aix Marseille Univ, INSERM, TAGC, Theory and Approaches of Genomic Complexity, UMR_S 1090, Marseille, France.
Aix Marseille Univ, CNRS, INSERM, CIML, Marseille, France.
Nucleic Acids Res. 2017 Jul 27;45(13):e119. doi: 10.1093/nar/gkx314.
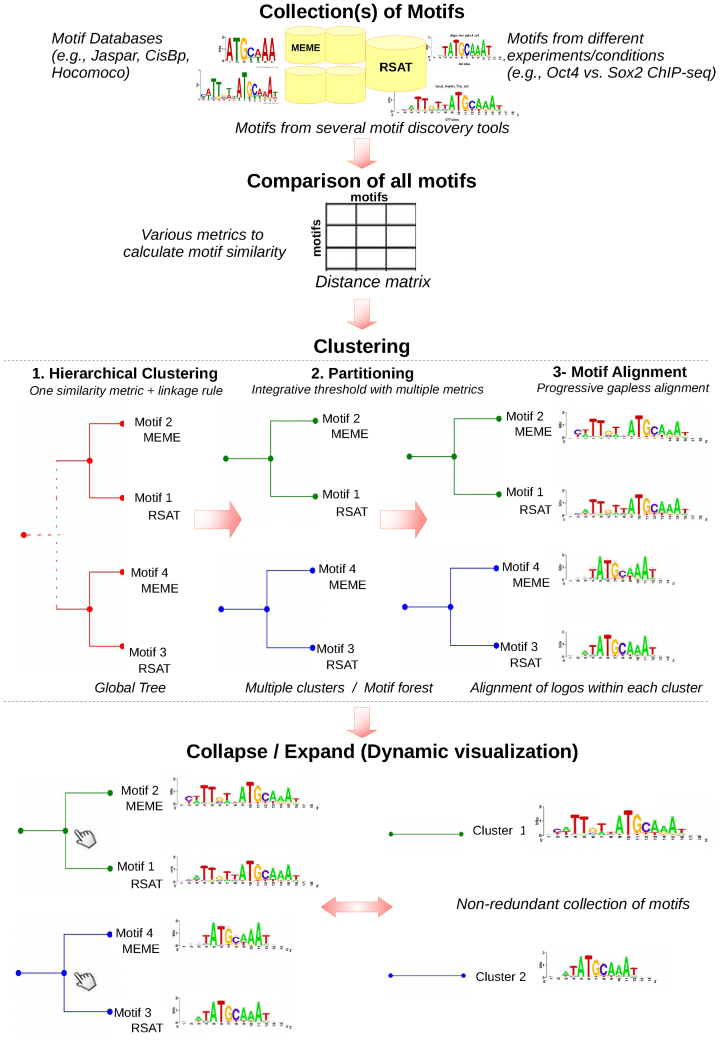
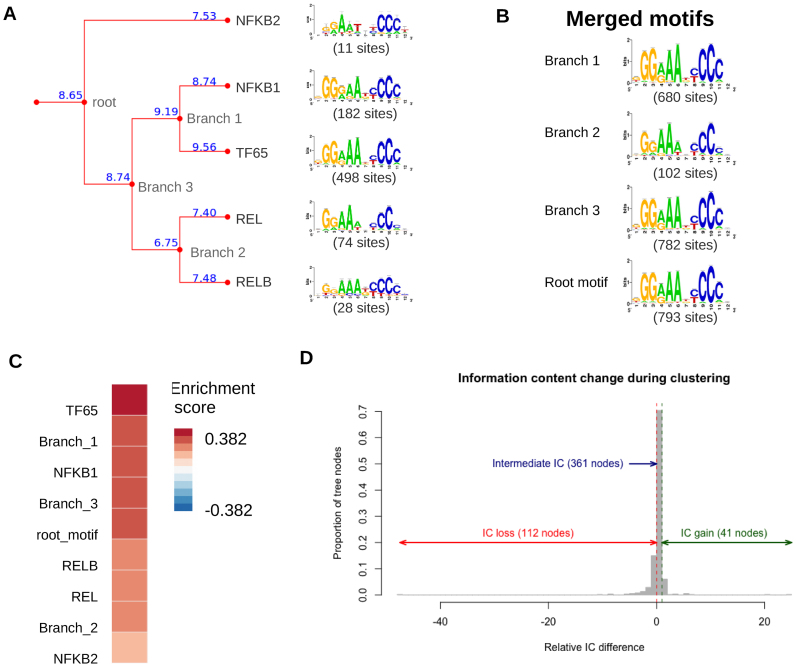
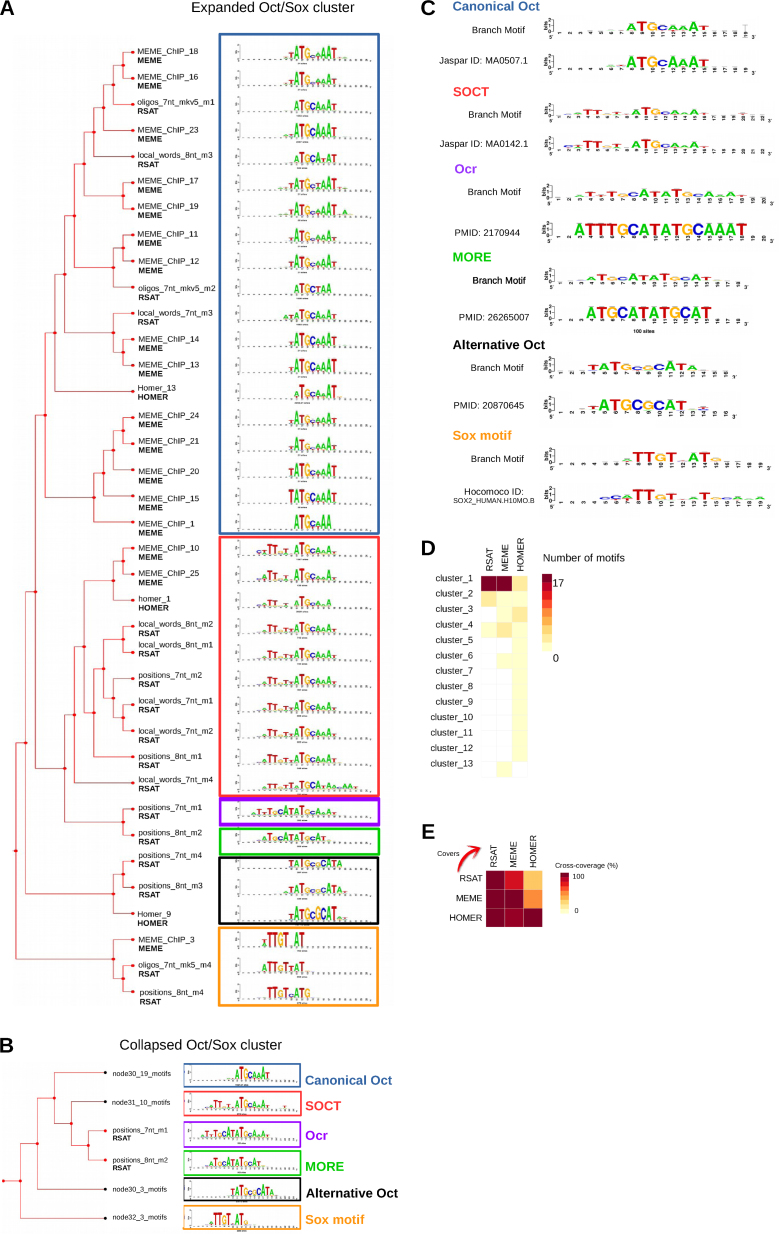
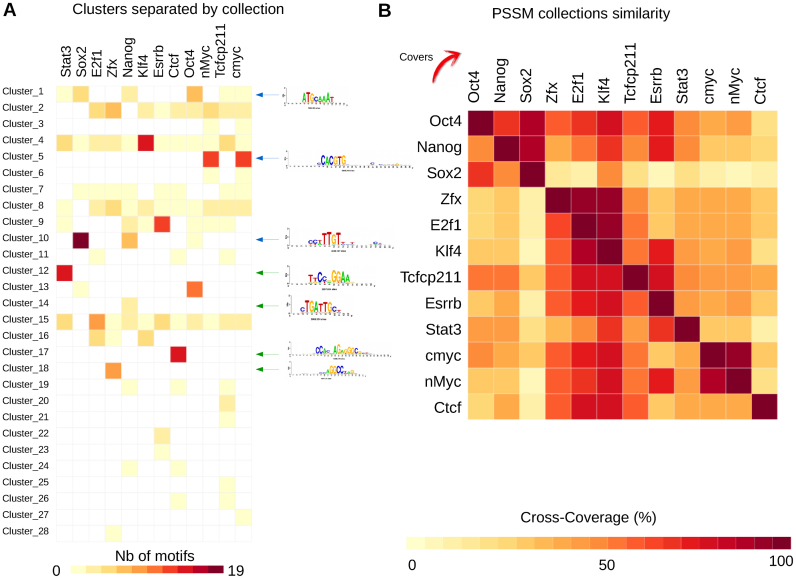
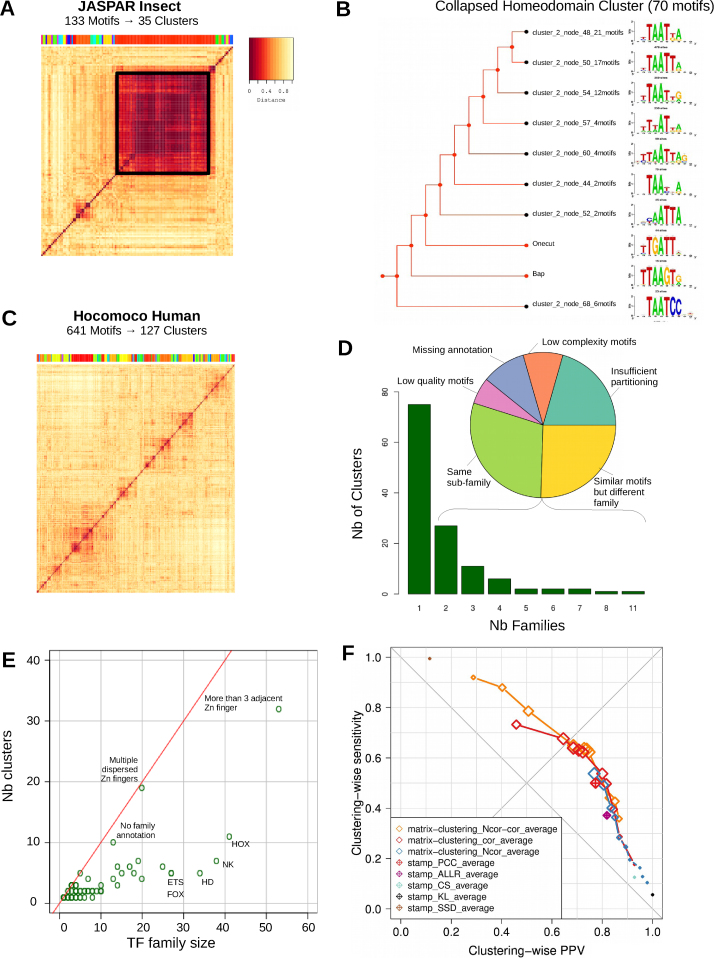
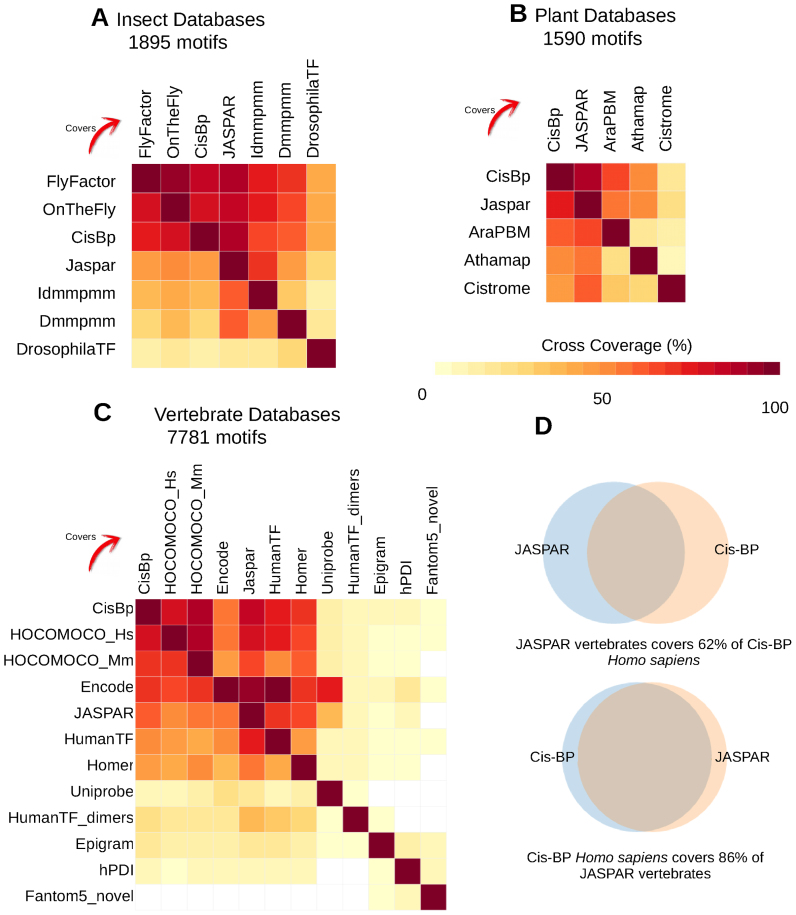
Transcription factor (TF) databases contain multitudes of binding motifs (TFBMs) from various sources, from which non-redundant collections are derived by manual curation. The advent of high-throughput methods stimulated the production of novel collections with increasing numbers of motifs. Meta-databases, built by merging these collections, contain redundant versions, because available tools are not suited to automatically identify and explore biologically relevant clusters among thousands of motifs. Motif discovery from genome-scale data sets (e.g. ChIP-seq) also produces redundant motifs, hampering the interpretation of results. We present matrix-clustering, a versatile tool that clusters similar TFBMs into multiple trees, and automatically creates non-redundant TFBM collections. A feature unique to matrix-clustering is its dynamic visualisation of aligned TFBMs, and its capability to simultaneously treat multiple collections from various sources. We demonstrate that matrix-clustering considerably simplifies the interpretation of combined results from multiple motif discovery tools, and highlights biologically relevant variations of similar motifs. We also ran a large-scale application to cluster ∼11 000 motifs from 24 entire databases, showing that matrix-clustering correctly groups motifs belonging to the same TF families, and drastically reduced motif redundancy. matrix-clustering is integrated within the RSAT suite (http://rsat.eu/), accessible through a user-friendly web interface or command-line for its integration in pipelines.
转录因子(TF)数据库包含来自各种来源的大量结合基序(TFBM),通过人工筛选从中获得非冗余集合。高通量方法的出现促使产生了包含越来越多基序的新集合。通过合并这些集合构建的元数据库包含冗余版本,因为现有的工具不适合自动识别和探索数千个基序中生物学相关的聚类。从基因组规模数据集(例如ChIP-seq)中发现基序也会产生冗余基序,这妨碍了对结果的解释。我们提出了矩阵聚类方法,这是一种通用工具,可将相似的TFBM聚类为多个树,并自动创建非冗余的TFBM集合。矩阵聚类的独特之处在于其对对齐的TFBM的动态可视化,以及同时处理来自各种来源的多个集合的能力。我们证明,矩阵聚类极大地简化了对来自多个基序发现工具的组合结果的解释,并突出了相似基序的生物学相关变异。我们还进行了大规模应用,对来自24个完整数据库的约11000个基序进行聚类,结果表明矩阵聚类正确地将属于同一TF家族的基序分组,并大幅减少了基序冗余。矩阵聚类集成在RSAT套件(http://rsat.eu/)中,可通过用户友好的网页界面或命令行访问,以便将其集成到工作流程中。