Institute of Machine Learning and Systems Biology, College of Electronics and Information Engineering, Tongji University, Shanghai, 201804, P.R. China.
Sci Rep. 2017 Jun 12;7(1):3217. doi: 10.1038/s41598-017-03554-7.
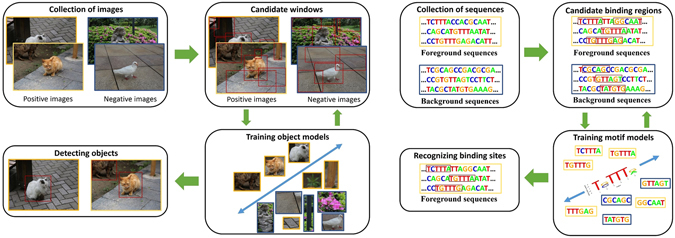
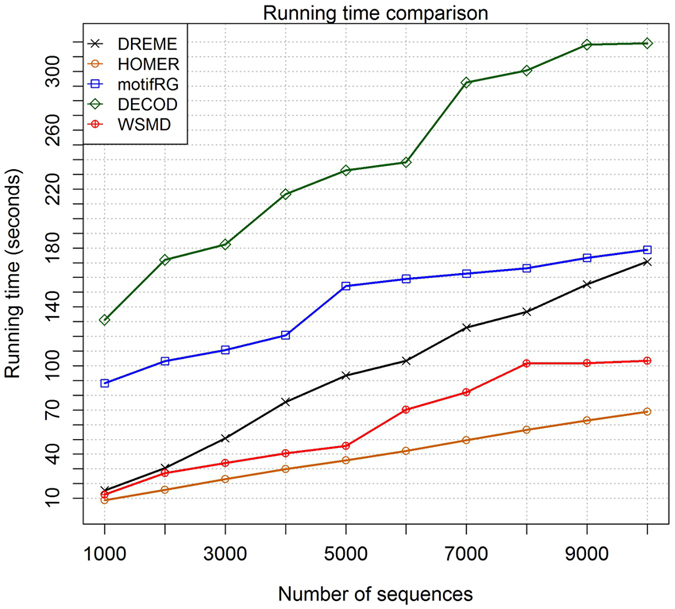
Although discriminative motif discovery (DMD) methods are promising for eliciting motifs from high-throughput experimental data, due to consideration of computational expense, most of existing DMD methods have to choose approximate schemes that greatly restrict the search space, leading to significant loss of predictive accuracy. In this paper, we propose Weakly-Supervised Motif Discovery (WSMD) to discover motifs from ChIP-seq datasets. In contrast to the learning strategies adopted by previous DMD methods, WSMD allows a "global" optimization scheme of the motif parameters in continuous space, thereby reducing the information loss of model representation and improving the quality of resultant motifs. Meanwhile, by exploiting the connection between DMD framework and existing weakly supervised learning (WSL) technologies, we also present highly scalable learning strategies for the proposed method. The experimental results on both real ChIP-seq datasets and synthetic datasets show that WSMD substantially outperforms former DMD methods (including DREME, HOMER, XXmotif, motifRG and DECOD) in terms of predictive accuracy, while also achieving a competitive computational speed.
尽管判别基序发现(DMD)方法在从高通量实验数据中提取基序方面很有前景,但由于考虑到计算费用,大多数现有的 DMD 方法不得不选择近似方案,这极大地限制了搜索空间,导致预测准确性的显著损失。在本文中,我们提出了弱监督基序发现(WSMD)来从 ChIP-seq 数据集中发现基序。与之前 DMD 方法采用的学习策略不同,WSMD 允许在连续空间中对基序参数进行“全局”优化方案,从而减少模型表示的信息损失,并提高所得基序的质量。同时,通过利用 DMD 框架和现有的弱监督学习(WSL)技术之间的联系,我们还为所提出的方法提供了高度可扩展的学习策略。在真实的 ChIP-seq 数据集和合成数据集上的实验结果表明,WSMD 在预测准确性方面明显优于以前的 DMD 方法(包括 DREME、HOMER、XXmotif、motifRG 和 DECOD),同时也实现了具有竞争力的计算速度。