Department of Neuroscience, International School for Advanced Studies (SISSA), Trieste, Italy.
Cell Death Dis. 2017 Jun 15;8(6):e2881. doi: 10.1038/cddis.2017.232.
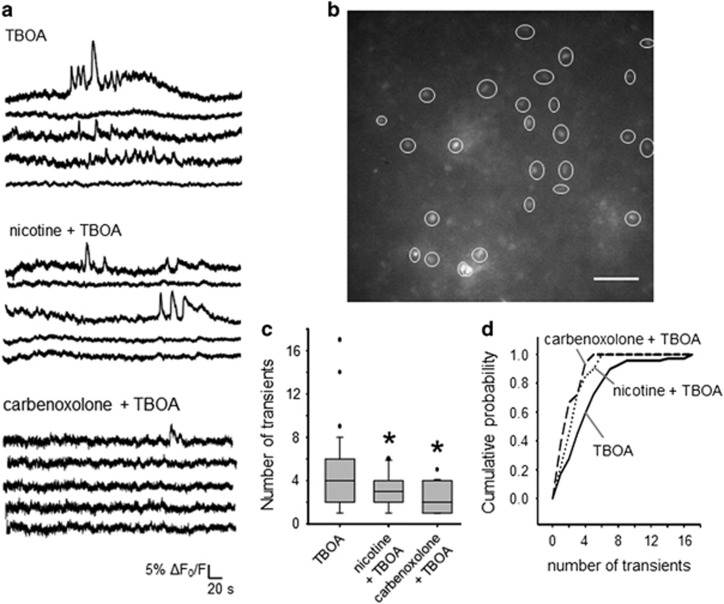
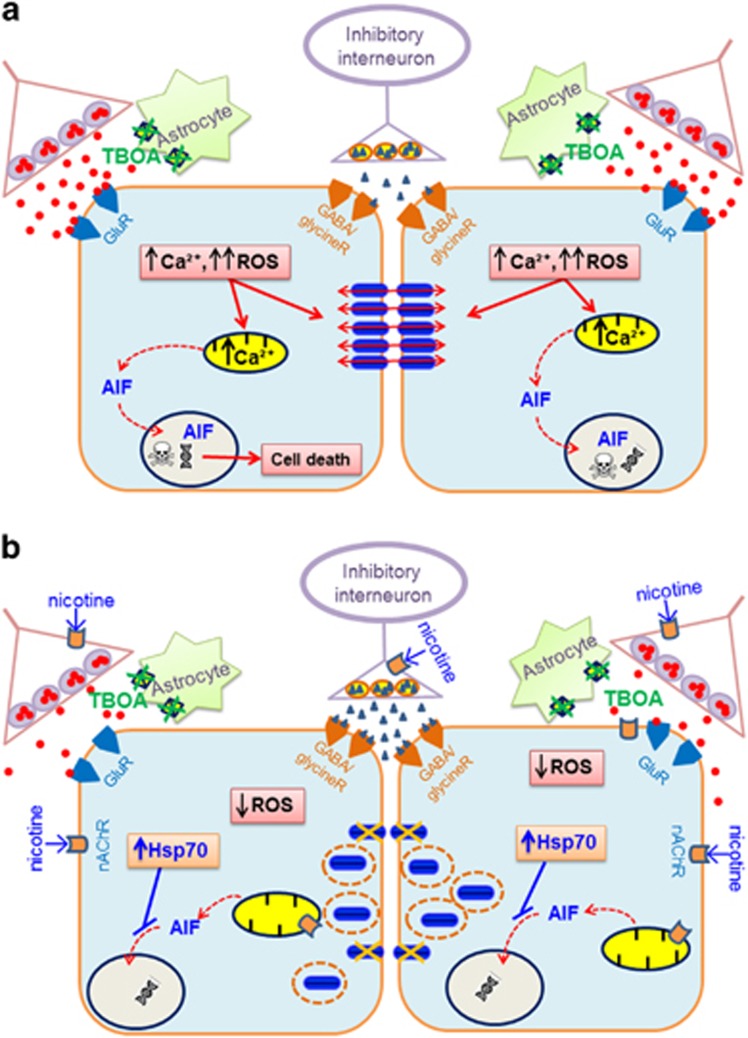
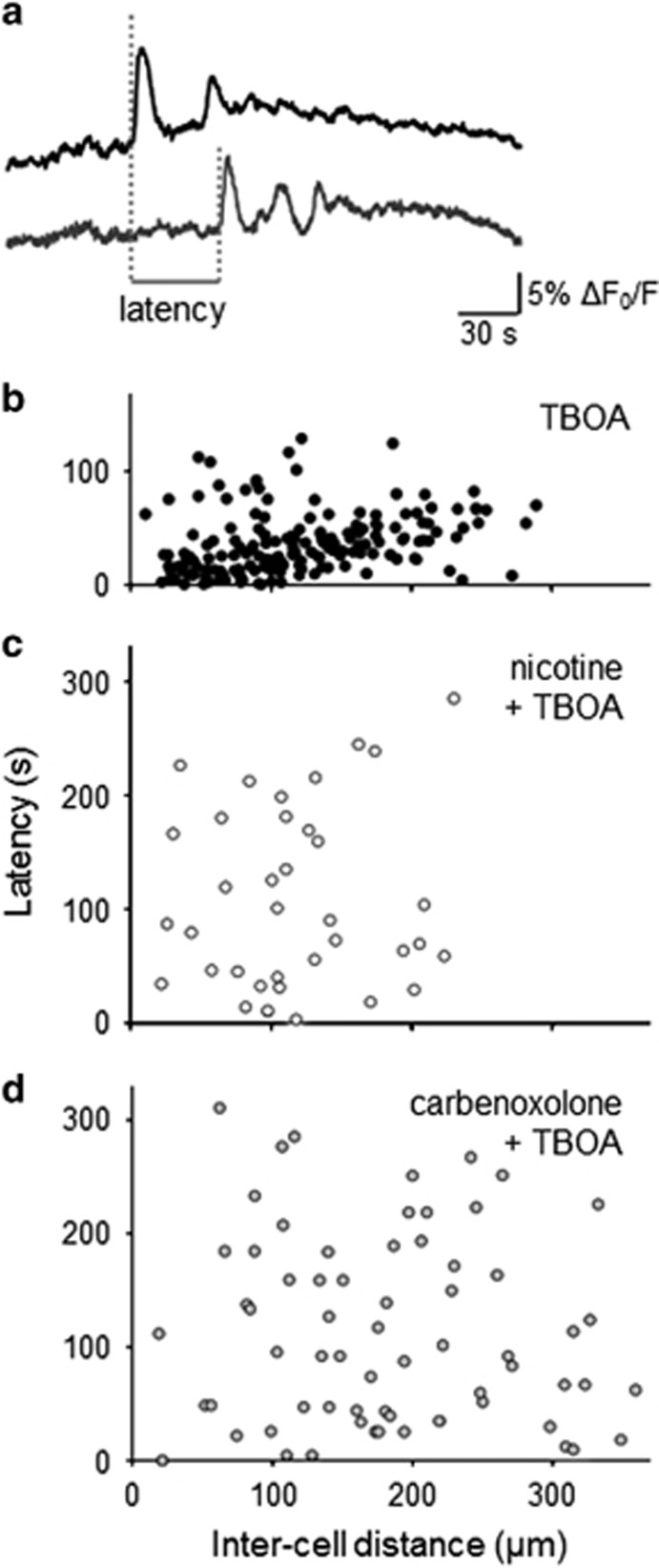
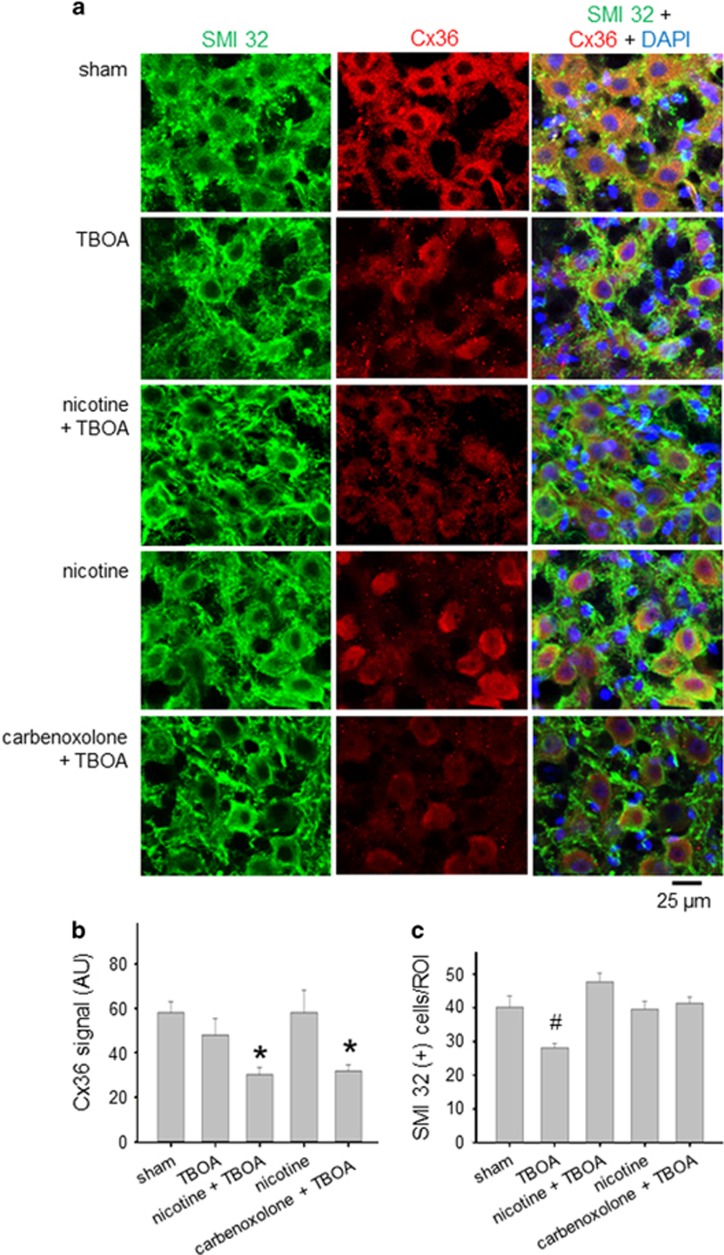
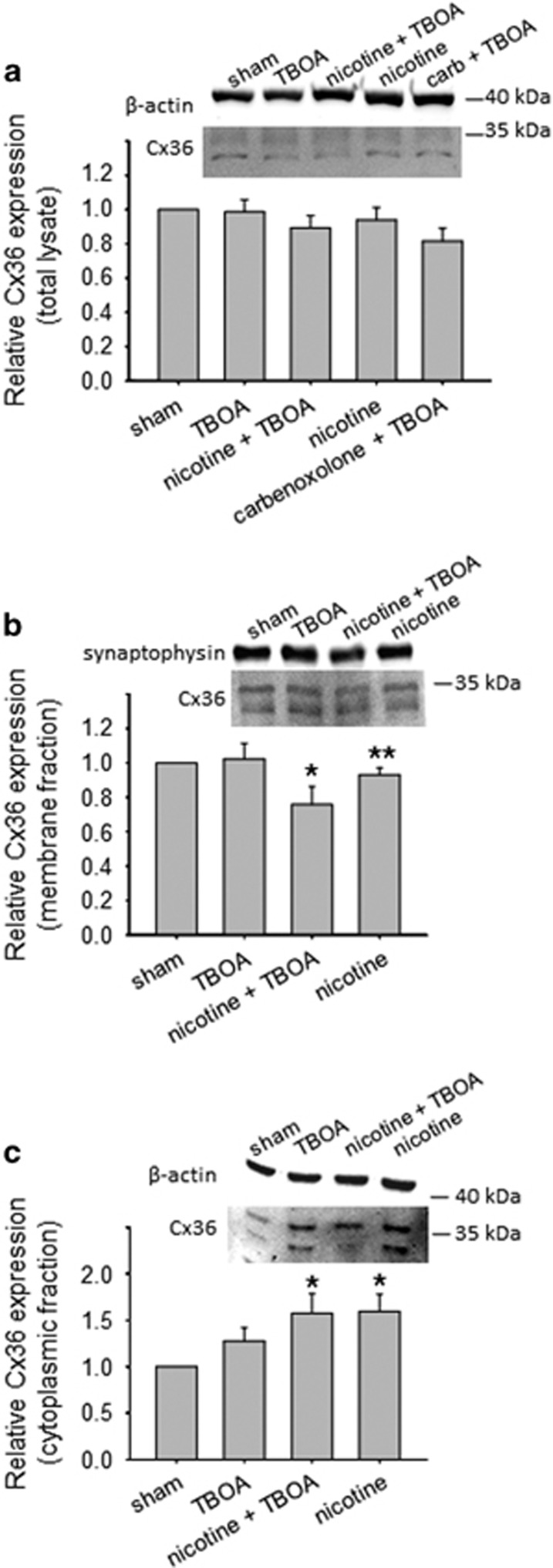
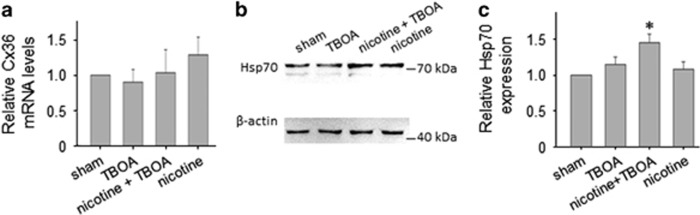
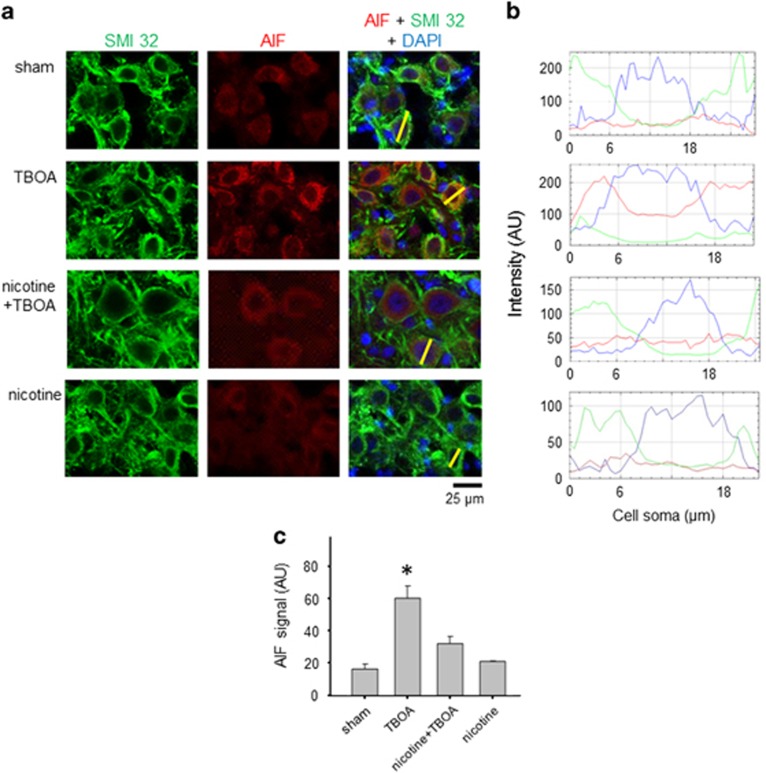
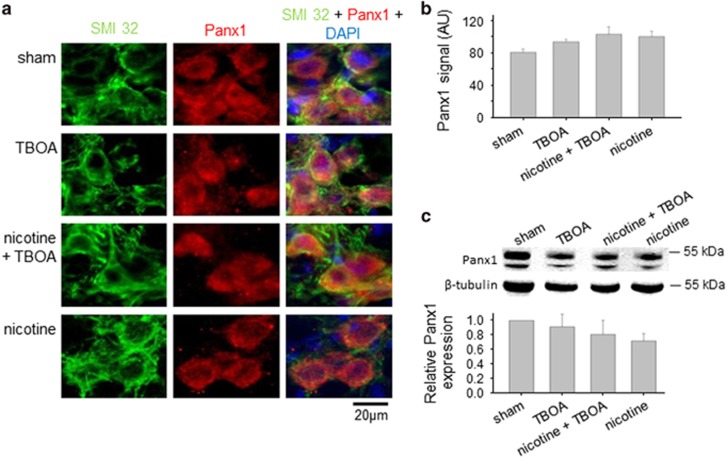
Motoneuron disease including amyotrophic lateral sclerosis may be due, at an early stage, to deficit in the extracellular clearance of the excitatory transmitter glutamate. A model of glutamate-mediated excitotoxic cell death based on pharmacological inhibition of its uptake was used to investigate how activation of neuronal nicotinic receptors by nicotine may protect motoneurons. Hypoglossal motoneurons (HMs) in neonatal rat brainstem slices were exposed to the glutamate uptake blocker DL-threo-β-benzyloxyaspartate (TBOA) that evoked large Ca transients time locked among nearby HMs, whose number fell by about 30% 4 h later. As nicotine or the gap junction blocker carbenoxolone suppressed bursting, we studied connexin 36 (Cx36), which constitutes gap junctions in neurons and found it largely expressed by HMs. Cx36 was downregulated when nicotine or carbenoxolone was co-applied with TBOA. Expression of Cx36 was preferentially observed in cytosolic rather than membrane fractions after nicotine and TBOA, suggesting protein redistribution with no change in synthesis. Nicotine raised the expression of heat shock protein 70 (Hsp70), a protective factor that binds the apoptotic-inducing factor (AIF) whose nuclear translocation is a cause of cell death. TBOA increased intracellular AIF, an effect blocked by nicotine. These results indicate that activation of neuronal nicotinic receptors is an early tool for protecting motoneurons from excitotoxicity and that this process is carried out via the combined decrease in Cx36 activity, overexpression of Hsp70 and fall in AIF translocation. Thus, retarding or inhibiting HM death may be experimentally achieved by targeting one of these processes leading to motoneuron death.
运动神经元疾病,包括肌萎缩侧索硬化症,可能在早期阶段由于兴奋性递质谷氨酸的细胞外清除缺陷所致。使用基于其摄取的药理学抑制的谷氨酸介导的兴奋性毒性细胞死亡模型来研究尼古丁如何通过激活神经元烟碱型受体来保护运动神经元。新生大鼠延髓脑片的舌下运动神经元(HMs)暴露于谷氨酸摄取阻滞剂 DL-threo-β-苄氧基天冬氨酸(TBOA),TBOA 引发附近 HMs 之间时间锁定的大 Ca 瞬变,4 小时后其数量下降约 30%。由于尼古丁或缝隙连接阻滞剂 carbenoxolone 抑制爆发,我们研究了构成神经元缝隙连接的连接蛋白 36(Cx36),并发现其主要由 HMs 表达。当尼古丁或 carbenoxolone 与 TBOA 一起应用时,Cx36 下调。尼古丁和 TBOA 后,Cx36 主要在细胞质而不是膜部分表达,表明蛋白重新分布而没有合成变化。尼古丁增加了热休克蛋白 70(Hsp70)的表达,Hsp70 是一种保护性因子,可结合凋亡诱导因子(AIF),AIF 的核易位是细胞死亡的原因。TBOA 增加了细胞内 AIF 的含量,尼古丁可阻止该作用。这些结果表明,神经元烟碱型受体的激活是保护运动神经元免受兴奋性毒性的早期工具,并且该过程通过 Cx36 活性的联合降低,Hsp70 的过表达和 AIF 易位的减少来进行。因此,通过针对导致运动神经元死亡的这些过程之一,延迟或抑制 HM 死亡可能在实验上得以实现。