Magnusson Rasmus, Mariotti Guido Pio, Köpsén Mattias, Lövfors William, Gawel Danuta R, Jörnsten Rebecka, Linde Jörg, Nordling Torbjörn E M, Nyman Elin, Schulze Sylvie, Nestor Colm E, Zhang Huan, Cedersund Gunnar, Benson Mikael, Tjärnberg Andreas, Gustafsson Mika
Bioinformatics Unit, Department of Physics, Chemistry and Biology, Linköping University, Linköping, Sweden.
Centre for Personalised Medicine, Department of Clinical and Experimental Medicine, Linköping University, Linköping, Sweden.
PLoS Comput Biol. 2017 Jun 22;13(6):e1005608. doi: 10.1371/journal.pcbi.1005608. eCollection 2017 Jun.
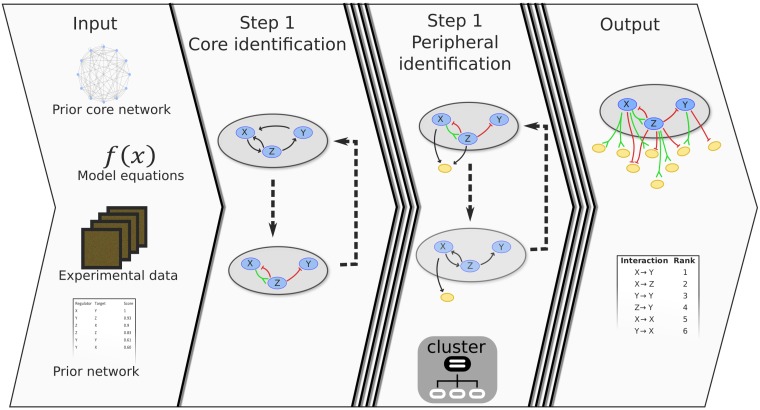
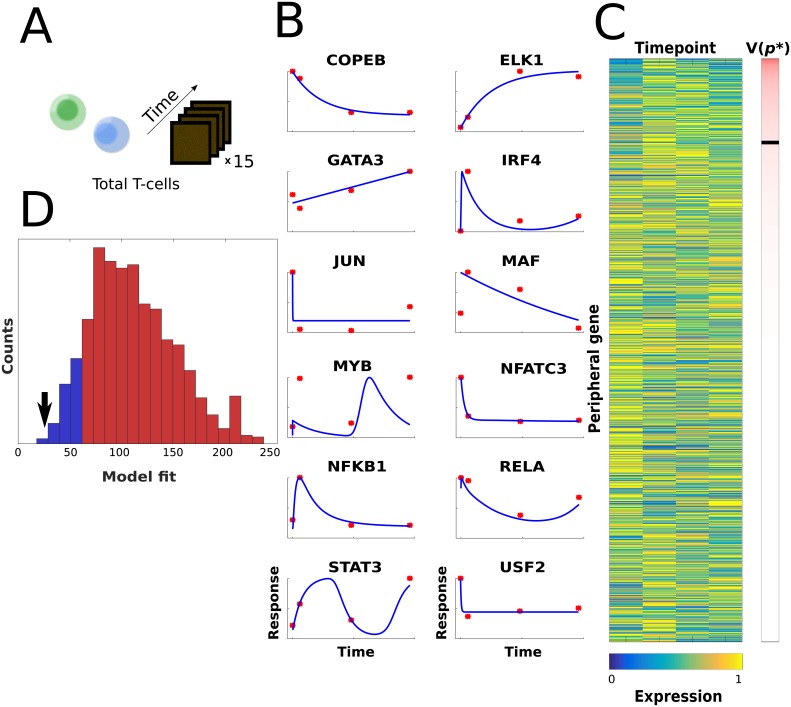
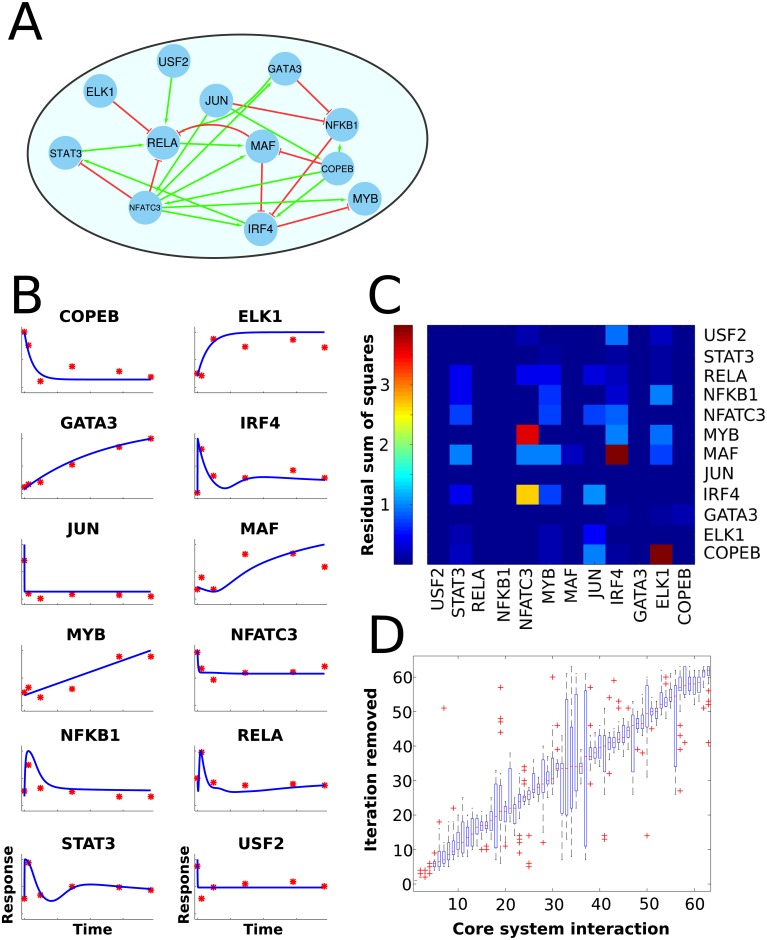
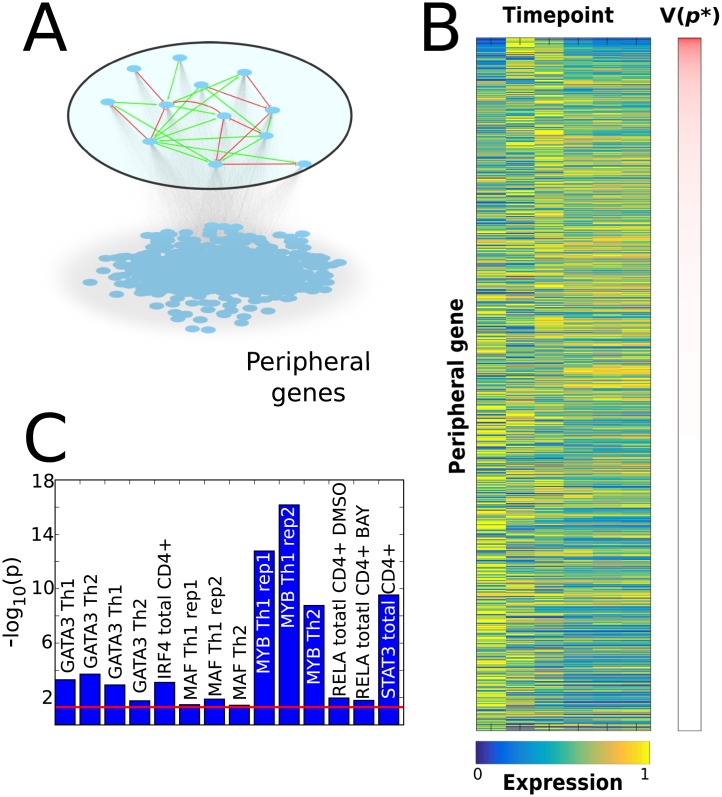
Recent technological advancements have made time-resolved, quantitative, multi-omics data available for many model systems, which could be integrated for systems pharmacokinetic use. Here, we present large-scale simulation modeling (LASSIM), which is a novel mathematical tool for performing large-scale inference using mechanistically defined ordinary differential equations (ODE) for gene regulatory networks (GRNs). LASSIM integrates structural knowledge about regulatory interactions and non-linear equations with multiple steady state and dynamic response expression datasets. The rationale behind LASSIM is that biological GRNs can be simplified using a limited subset of core genes that are assumed to regulate all other gene transcription events in the network. The LASSIM method is implemented as a general-purpose toolbox using the PyGMO Python package to make the most of multicore computers and high performance clusters, and is available at https://gitlab.com/Gustafsson-lab/lassim. As a method, LASSIM works in two steps, where it first infers a non-linear ODE system of the pre-specified core gene expression. Second, LASSIM in parallel optimizes the parameters that model the regulation of peripheral genes by core system genes. We showed the usefulness of this method by applying LASSIM to infer a large-scale non-linear model of naïve Th2 cell differentiation, made possible by integrating Th2 specific bindings, time-series together with six public and six novel siRNA-mediated knock-down experiments. ChIP-seq showed significant overlap for all tested transcription factors. Next, we performed novel time-series measurements of total T-cells during differentiation towards Th2 and verified that our LASSIM model could monitor those data significantly better than comparable models that used the same Th2 bindings. In summary, the LASSIM toolbox opens the door to a new type of model-based data analysis that combines the strengths of reliable mechanistic models with truly systems-level data. We demonstrate the power of this approach by inferring a mechanistically motivated, genome-wide model of the Th2 transcription regulatory system, which plays an important role in several immune related diseases.
最近的技术进步使得许多模型系统能够获得时间分辨的、定量的多组学数据,这些数据可用于系统药代动力学整合。在此,我们介绍大规模模拟建模(LASSIM),它是一种新颖的数学工具,用于使用针对基因调控网络(GRN)的机械定义的常微分方程(ODE)进行大规模推断。LASSIM将关于调控相互作用的结构知识和非线性方程与多个稳态和动态响应表达数据集进行整合。LASSIM背后的基本原理是,生物GRN可以使用有限的核心基因子集进行简化,这些核心基因被假定调控网络中的所有其他基因转录事件。LASSIM方法作为一个通用工具箱,使用PyGMO Python包来充分利用多核计算机和高性能集群,可在https://gitlab.com/Gustafsson-lab/lassim获取。作为一种方法,LASSIM分两步工作,首先推断预先指定的核心基因表达的非线性ODE系统。其次,LASSIM并行优化由核心系统基因对外周基因调控进行建模的参数。我们通过将LASSIM应用于推断幼稚Th2细胞分化的大规模非线性模型,展示了该方法的实用性,这通过整合Th2特异性结合、时间序列以及六个公共和六个新的siRNA介导的敲低实验得以实现。ChIP-seq显示所有测试转录因子均有显著重叠。接下来,我们在向Th2分化过程中对总T细胞进行了新的时间序列测量,并验证我们的LASSIM模型能够比使用相同Th2结合的可比模型更好地监测这些数据。总之,LASSIM工具箱为一种新型的基于模型的数据分析打开了大门,这种分析将可靠的机械模型的优势与真正的系统级数据相结合。我们通过推断一个在几种免疫相关疾病中起重要作用的Th2转录调控系统的机械驱动的全基因组模型,展示了这种方法的强大功能。