Mirra Virginia, Werner Claudius, Santamaria Francesca
Department of Translational Medical Sciences, Federico II University, Naples, Italy.
Department of Pediatrics, Federico II University, Naples, Italy.
Front Pediatr. 2017 Jun 9;5:135. doi: 10.3389/fped.2017.00135. eCollection 2017.
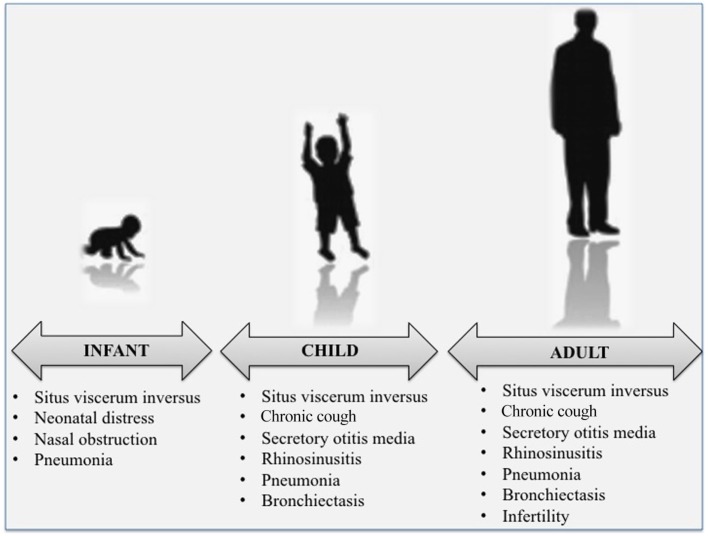
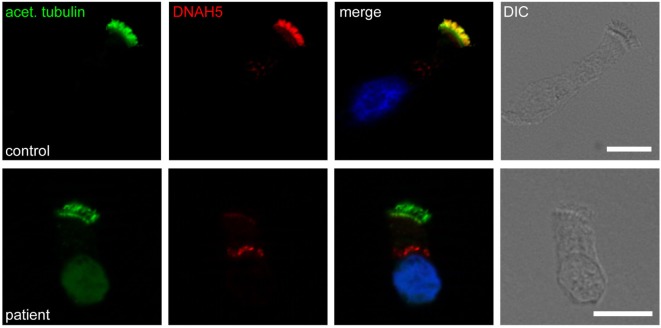
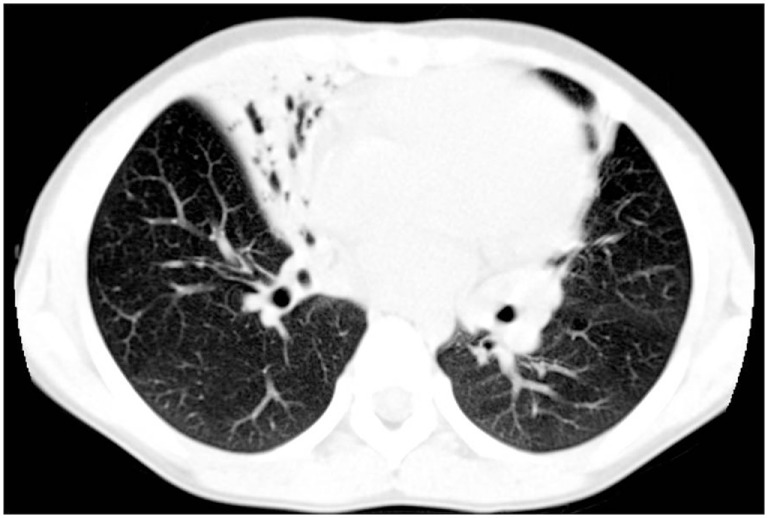
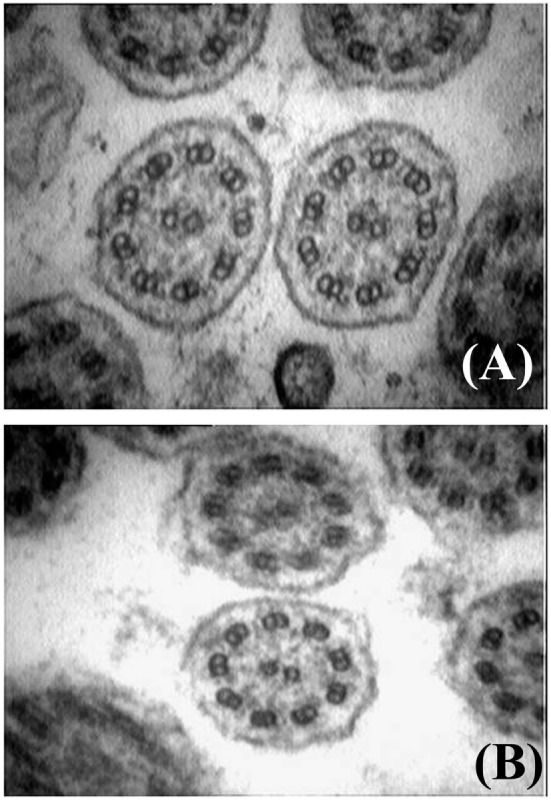
Primary ciliary dyskinesia (PCD) is an orphan disease (MIM 244400), autosomal recessive inherited, characterized by motile ciliary dysfunction. The estimated prevalence of PCD is 1:10,000 to 1:20,000 live-born children, but true prevalence could be even higher. PCD is characterized by chronic upper and lower respiratory tract disease, infertility/ectopic pregnancy, and situs anomalies, that occur in ≈50% of PCD patients (Kartagener syndrome), and these may be associated with congenital heart abnormalities. Most patients report a daily year-round wet cough or nose congestion starting in the first year of life. Daily wet cough, associated with recurrent infections exacerbations, results in the development of chronic suppurative lung disease, with localized-to-diffuse bronchiectasis. No diagnostic test is perfect for confirming PCD. Diagnosis can be challenging and relies on a combination of clinical data, nasal nitric oxide levels plus cilia ultrastructure and function analysis. Adjunctive tests include genetic analysis and repeated tests in ciliary culture specimens. There are currently 33 known genes associated with PCD and correlations between genotype and ultrastructural defects have been increasingly demonstrated. Comprehensive genetic testing may hopefully screen young infants before symptoms occur, thus improving survival. Recent surprising advances in PCD genetic designed a novel approach called "gene editing" to restore gene function and normalize ciliary motility, opening up new avenues for treating PCD. Currently, there are no data from randomized clinical trials to support any specific treatment, thus, management strategies are usually extrapolated from cystic fibrosis. The goal of treatment is to prevent exacerbations, slowing the progression of lung disease. The therapeutic mainstay includes airway clearance maneuvers mainly with nebulized hypertonic saline and chest physiotherapy, and prompt and aggressive administration of antibiotics. Standardized care at specialized centers using a multidisciplinary approach that imposes surveillance of lung function and of airway biofilm composition likely improves patients' outcome. Pediatricians, neonatologists, pulmonologists, and ENT surgeons should maintain high awareness of PCD and refer patients to the specialized center before sustained irreversible lung damage develops. The recent creation of a network of PCD clinical centers, focusing on improving diagnosis and treatment, will hopefully help to improve care and knowledge of PCD patients.
原发性纤毛运动障碍(PCD)是一种罕见病(MIM 244400),呈常染色体隐性遗传,其特征为纤毛运动功能障碍。据估计,PCD在活产儿中的患病率为1:10000至1:20000,但实际患病率可能更高。PCD的特征是慢性上、下呼吸道疾病、不孕/异位妊娠和内脏反位异常,约50%的PCD患者会出现这些症状(卡塔格内综合征),且这些症状可能与先天性心脏异常有关。大多数患者报告从出生第一年起就全年每日有湿性咳嗽或鼻塞。每日湿性咳嗽伴有反复感染加重,会导致慢性化脓性肺病,并伴有局限性至弥漫性支气管扩张。没有一种诊断测试能完美确诊PCD。诊断具有挑战性,依赖于临床数据、鼻一氧化氮水平以及纤毛超微结构和功能分析的综合判断。辅助检查包括基因分析和对纤毛培养标本的重复检测。目前已知有33个基因与PCD相关,且基因型与超微结构缺陷之间的相关性已得到越来越多的证实。全面的基因检测有望在症状出现前筛查出婴幼儿,从而提高生存率。PCD遗传学领域最近令人惊讶的进展设计出了一种名为“基因编辑”的新方法来恢复基因功能并使纤毛运动正常化,为治疗PCD开辟了新途径。目前,尚无随机临床试验数据支持任何特定治疗方法,因此,管理策略通常是从囊性纤维化治疗中推断而来。治疗的目标是预防病情加重,减缓肺部疾病的进展。治疗的主要手段包括主要采用雾化高渗盐水和胸部物理治疗的气道清理措施,以及及时、积极地使用抗生素。在专业中心采用多学科方法进行标准化护理,对肺功能和气道生物膜成分进行监测,可能会改善患者的预后。儿科医生、新生儿科医生、肺科医生和耳鼻喉科外科医生应高度重视PCD,并在持续性不可逆肺损伤发生前将患者转诊至专业中心。最近建立的PCD临床中心网络专注于改善诊断和治疗,有望帮助提高对PCD患者的护理水平和认知。