David Sophia, Sánchez-Busó Leonor, Harris Simon R, Marttinen Pekka, Rusniok Christophe, Buchrieser Carmen, Harrison Timothy G, Parkhill Julian
Pathogen Genomics, Wellcome Trust Sanger Institute, Cambridge, United Kingdom.
Respiratory and Vaccine Preventable Bacteria Reference Unit, Public Health England, London, United Kingdom.
PLoS Genet. 2017 Jun 26;13(6):e1006855. doi: 10.1371/journal.pgen.1006855. eCollection 2017 Jun.
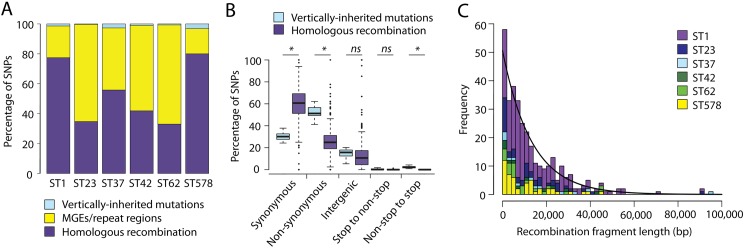
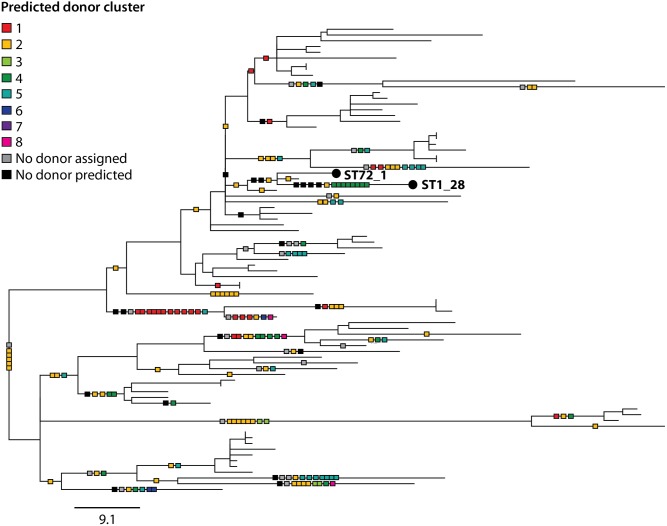
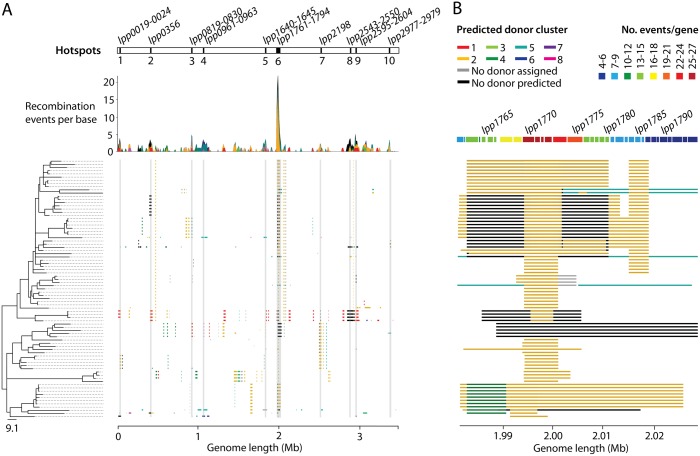
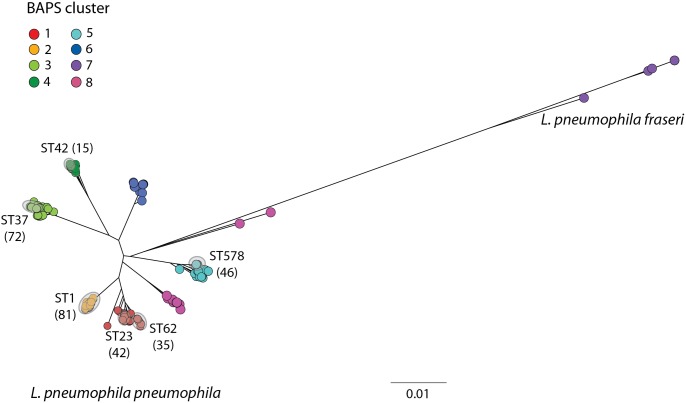
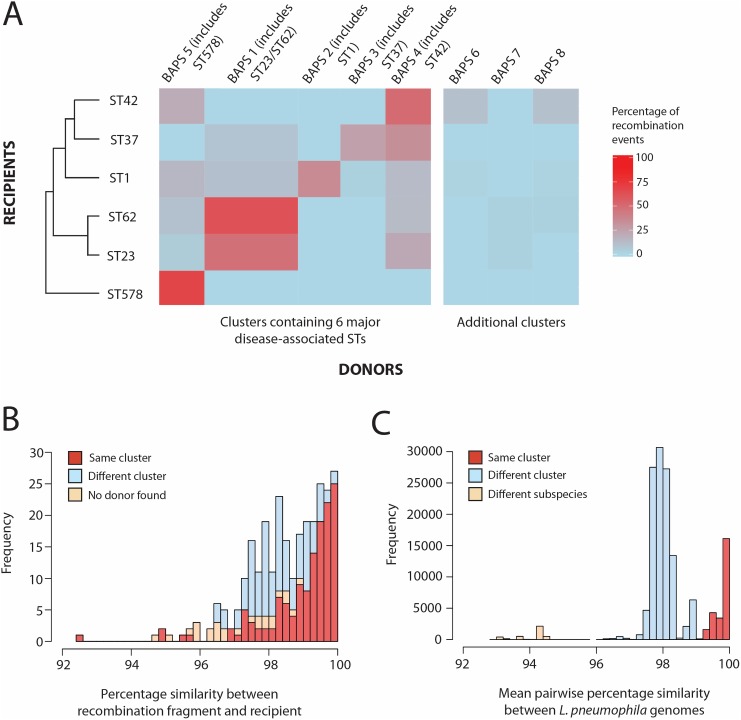
Legionella pneumophila is an environmental bacterium and the causative agent of Legionnaires' disease. Previous genomic studies have shown that recombination accounts for a high proportion (>96%) of diversity within several major disease-associated sequence types (STs) of L. pneumophila. This suggests that recombination represents a potentially important force shaping adaptation and virulence. Despite this, little is known about the biological effects of recombination in L. pneumophila, particularly with regards to homologous recombination (whereby genes are replaced with alternative allelic variants). Using newly available population genomic data, we have disentangled events arising from homologous and non-homologous recombination in six major disease-associated STs of L. pneumophila (subsp. pneumophila), and subsequently performed a detailed characterisation of the dynamics and impact of homologous recombination. We identified genomic "hotspots" of homologous recombination that include regions containing outer membrane proteins, the lipopolysaccharide (LPS) region and Dot/Icm effectors, which provide interesting clues to the selection pressures faced by L. pneumophila. Inference of the origin of the recombined regions showed that isolates have most frequently imported DNA from isolates belonging to their own clade, but also occasionally from other major clades of the same subspecies. This supports the hypothesis that the possibility for horizontal exchange of new adaptations between major clades of the subspecies may have been a critical factor in the recent emergence of several clinically important STs from diverse genomic backgrounds. However, acquisition of recombined regions from another subspecies, L. pneumophila subsp. fraseri, was rarely observed, suggesting the existence of a recombination barrier and/or the possibility of ongoing speciation between the two subspecies. Finally, we suggest that multi-fragment recombination may occur in L. pneumophila, whereby multiple non-contiguous segments that originate from the same molecule of donor DNA are imported into a recipient genome during a single episode of recombination.
嗜肺军团菌是一种环境细菌,也是军团病的病原体。先前的基因组研究表明,重组在嗜肺军团菌几种主要疾病相关序列类型(STs)的多样性中占很高比例(>96%)。这表明重组是塑造适应性和毒力的潜在重要力量。尽管如此,关于嗜肺军团菌中重组的生物学效应,尤其是同源重组(即基因被替代等位基因变体取代)的生物学效应,人们了解甚少。利用新获得的群体基因组数据,我们解析了嗜肺军团菌(嗜肺军团菌亚种)六个主要疾病相关STs中同源和非同源重组产生的事件,并随后对同源重组的动态和影响进行了详细表征。我们确定了同源重组的基因组“热点”,包括含有外膜蛋白、脂多糖(LPS)区域和Dot/Icm效应器的区域,这为嗜肺军团菌面临的选择压力提供了有趣线索。对重组区域起源的推断表明,分离株最常从属于其自身进化枝的分离株中导入DNA,但偶尔也会从同一亚种的其他主要进化枝中导入。这支持了这样一种假设,即亚种主要进化枝之间新适应性水平交换的可能性可能是最近从不同基因组背景中出现几种临床重要STs的关键因素。然而,很少观察到从另一个亚种嗜肺军团菌弗雷泽亚种获得重组区域,这表明存在重组障碍和/或两个亚种之间正在进行物种形成的可能性。最后,我们认为嗜肺军团菌可能发生多片段重组,即在单次重组事件中,来自同一供体DNA分子的多个不连续片段被导入受体基因组。