Inlora Jingga, Sailani M Reza, Khodadadi Hamidreza, Teymurinezhad Ahmad, Takahashi Shinichi, Bernstein Jonathan A, Garshasbi Masoud, Snyder Michael P
Department of Genetics, Stanford University, Stanford, California 94305, USA.
Department of Medical Genetics, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran 19839-63113, Iran.
Cold Spring Harb Mol Case Stud. 2017 Nov 21;3(6). doi: 10.1101/mcs.a002014. Print 2017 Nov.
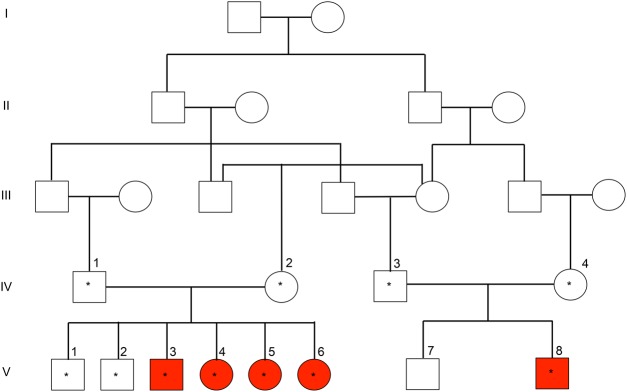
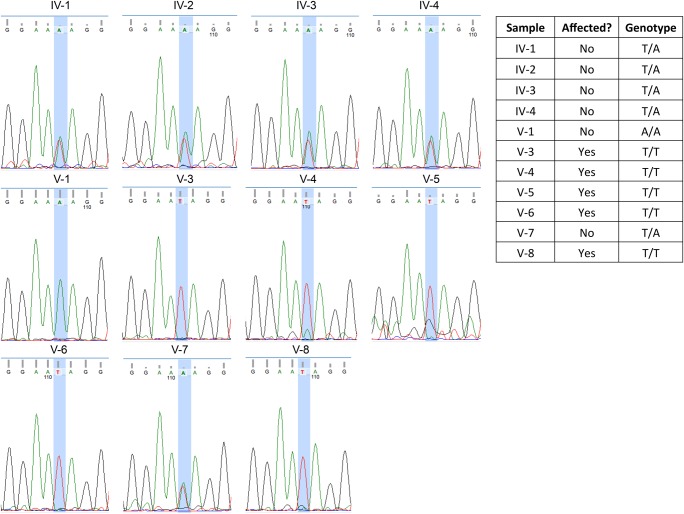
Hereditary ataxias are a clinically and genetically heterogeneous family of disorders defined by the inability to control gait and muscle coordination. Given the nonspecific symptoms of many hereditary ataxias, precise diagnosis relies on molecular genetic testing. To this end, we conducted whole-exome sequencing (WES) on a large consanguineous Iranian family with hereditary ataxia and oculomotor apraxia. WES in five affected and six unaffected individuals resulted in the identification of a homozygous novel stop-gain mutation in the gene (c.739A>T; p.Lys247*) that segregates with the phenotype. Mutations in the (OMIM 606350) gene are associated with ataxia with oculomotor apraxia type 1 (OMIM 208920).
遗传性共济失调是一类临床和遗传异质性疾病,其定义为无法控制步态和肌肉协调。鉴于许多遗传性共济失调的症状不具特异性,精确诊断依赖于分子基因检测。为此,我们对一个患有遗传性共济失调和动眼性失用症的伊朗近亲大家族进行了全外显子组测序(WES)。对5名患者和6名未患病个体进行的WES检测,发现该基因存在一个纯合的新型终止密码子获得性突变(c.739A>T;p.Lys247*),该突变与表型共分离。该(OMIM 606350)基因的突变与1型动眼性失用症伴共济失调(OMIM 208920)相关。