Ataxia Unit, Department of Neurology, Universidade Federal de São Paulo, Sao Paulo, BR.
Movement Disorders Unit, Departament of Internal Medicine, Universidade Federal de Juiz de Fora, Juiz de Fora, BR.
Tremor Other Hyperkinet Mov (N Y). 2020 Oct 7;10:39. doi: 10.5334/tohm.557.
Ataxia with oculomotor apraxia (AOA1) is characterized by early-onset progressive cerebellar ataxia with peripheral neuropathy, oculomotor apraxia and hypoalbuminemia and hypercholesterolemia.
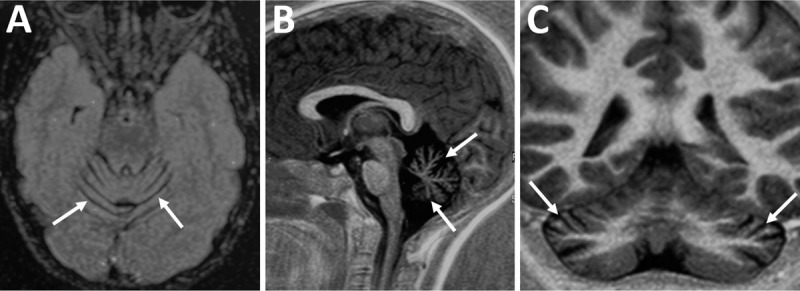
A 23-year-old previously healthy woman presented with slowly-progressive gait impairment since the age of six years. Neurological examination revealed profound areflexia, chorea, generalized dystonia and oculomotor apraxia. Brain MRI revealed mild cerebellar atrophy and needle EMG showed axonal sensorimotor neuropathy. Whole exome sequencing revealed a mutation in the aprataxin gene.
AOA1 can present with choreoathetosis mixed with dystonic features, resembling ataxia-telangiectasia. This case is instructive since mixed and complex movement disorders is not very common in AOA1.
Ataxia with oculomotor apraxia type 1 (AOA1) is characterized by early-onset ataxia and oculomotor apraxia caused by variants in the APTX gene.Ataxia is usually not the sole movement abnormality in AOA1.Hyperkinetic movement disorders, especially chorea and dystonia, may occur.Mixed and complex movement disorders is not very common in AOA1.Patients with early-onset ataxia associated with mixed movement disorders should also be investigated for AOA1.
眼动性运动不能症伴共济失调(AOA1)的特征是早期发病的进行性小脑共济失调伴周围神经病、眼动性运动不能和低白蛋白血症及高胆固醇血症。
一名 23 岁既往健康的女性,自 6 岁起出现缓慢进展的步态障碍。神经检查显示明显的反射消失、舞蹈症、全身性肌张力障碍和眼动性运动不能。脑部 MRI 显示轻度小脑萎缩,针极肌电图显示轴索性感觉运动神经病。全外显子组测序显示 aprataxin 基因突变。
AOA1 可表现为舞蹈-手足徐动症伴张力障碍特征,类似于共济失调-毛细血管扩张症。本病例具有指导意义,因为 AOA1 中混合和复杂运动障碍并不常见。
眼动性运动不能症伴共济失调 1 型(AOA1)的特征是由 APTX 基因突变引起的早期发病的共济失调和眼动性运动不能。在 AOA1 中,共济失调通常不是唯一的运动异常。可能出现运动过度障碍,特别是舞蹈症和肌张力障碍。混合和复杂运动障碍在 AOA1 中并不常见。对于伴有混合运动障碍的早发性共济失调患者,也应进行 AOA1 的检查。