Machado Henrique, Gram Lone
Department of Biotechnology and Biomedicine, Technical University of Denmark, MatematiktorvetKgs Lyngby, Denmark.
Novo Nordisk Foundation Center for Biosustainability, Technical University of DenmarkHørsholm, Denmark.
Front Microbiol. 2017 Jun 29;8:1204. doi: 10.3389/fmicb.2017.01204. eCollection 2017.
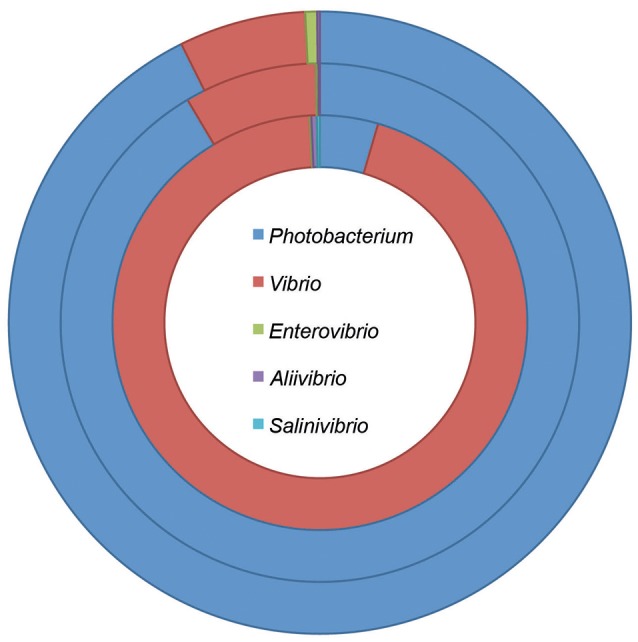
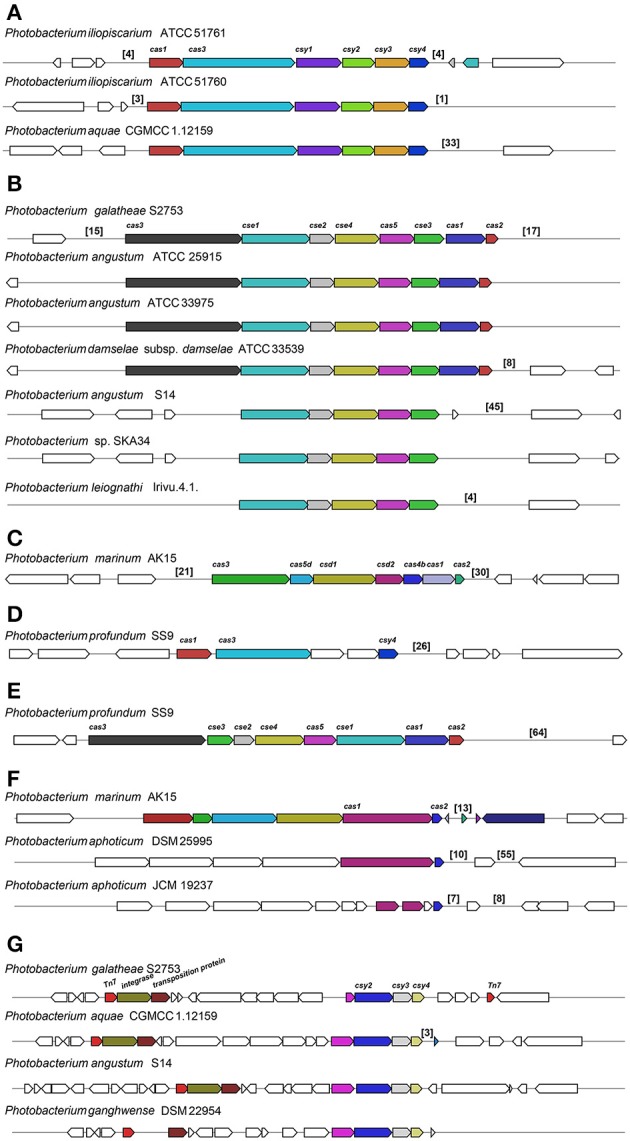
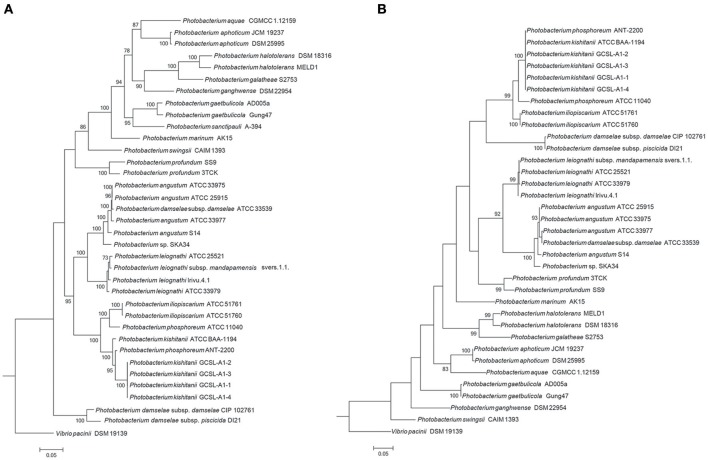
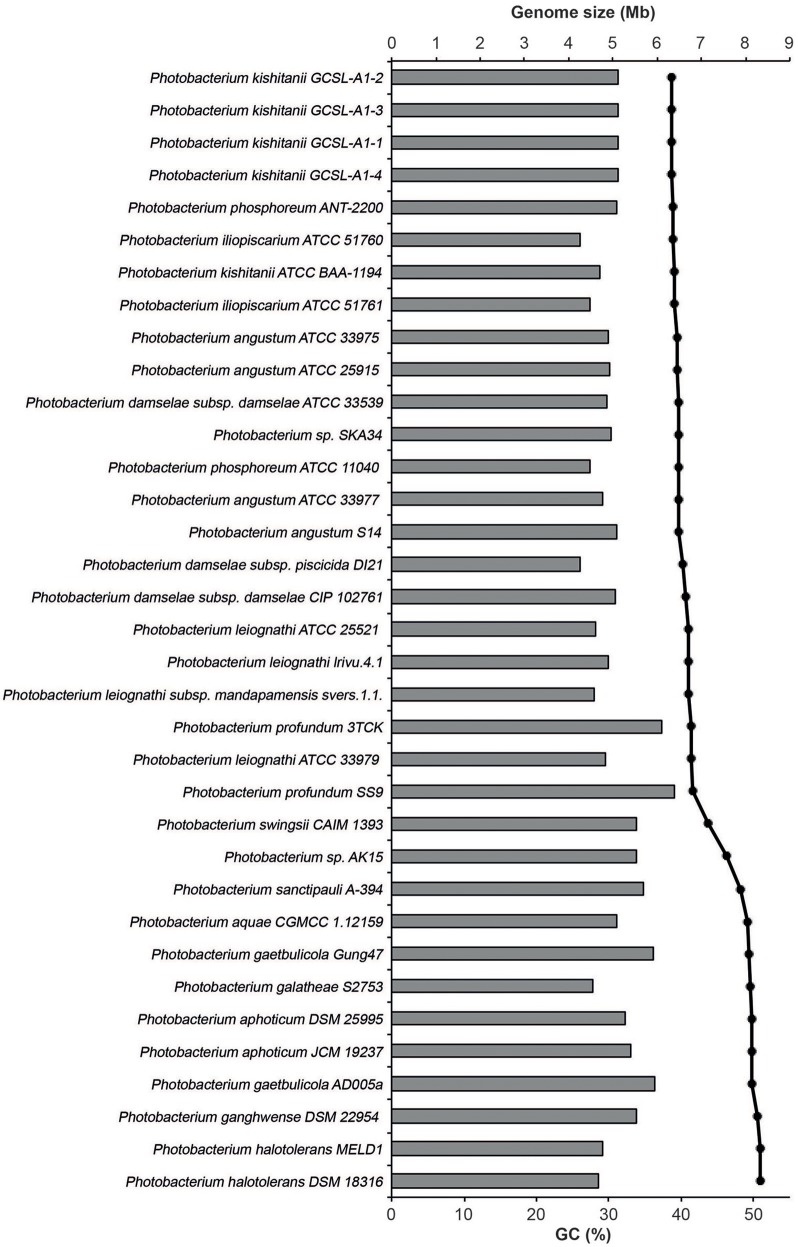
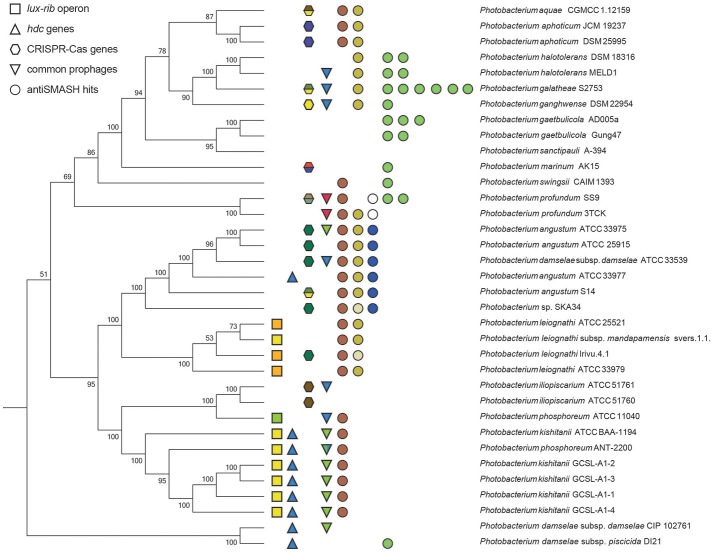
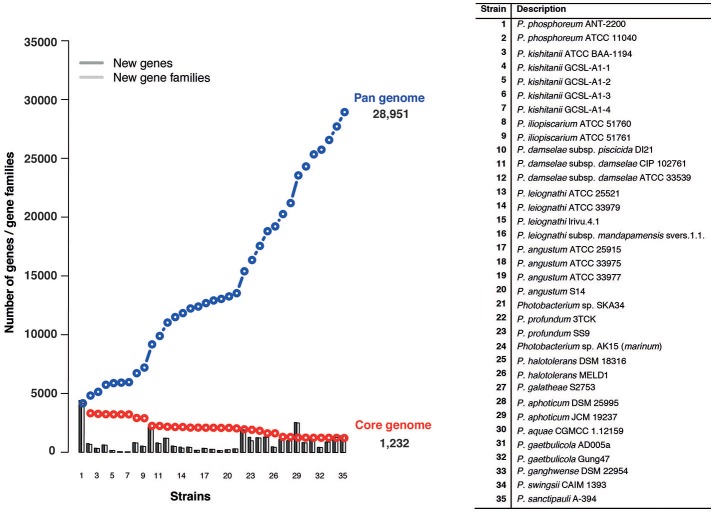
is a large marine bacterial family, which can constitute up to 50% of the prokaryotic population in marine waters. is the second largest genus in the family and we used comparative genomics on 35 strains representing 16 of the 28 species described so far, to understand the genomic diversity present in the genus. Such understanding is important for ecophysiology studies of the genus. We used whole genome sequences to evaluate phylogenetic relationships using several analyses (16S rRNA, MLSA, , amino-acid usage, ANI), which allowed us to identify two misidentified strains. Genome analyses also revealed occurrence of higher and lower GC content clades, correlating with phylogenetic clusters. Pan- and core-genome analysis revealed the conservation of 25% of the genome throughout the genus, with a large and open pan-genome. The major source of genomic diversity could be traced to the smaller chromosome and plasmids. Several of the physiological traits studied in the genus did not correlate with phylogenetic data. Since horizontal gene transfer (HGT) is often suggested as a source of genetic diversity and a potential driver of genomic evolution in bacterial species, we looked into evidence of such in genomes. Genomic islands were the source of genomic differences between strains of the same species. Also, we found transposase genes and CRISPR arrays that suggest multiple encounters with foreign DNA. Presence of genomic exchange traits was widespread and abundant in the genus, suggesting a role in genomic evolution. The high genetic variability and indications of genetic exchange make it difficult to elucidate genome evolutionary paths and raise the awareness of the roles of foreign DNA in the genomic evolution of environmental organisms.
是一个大型海洋细菌家族,在海水中可构成高达50%的原核生物种群。是该家族中第二大属,我们对代表迄今为止描述的28个物种中的16个的35个菌株进行了比较基因组学研究,以了解该属中存在的基因组多样性。这种理解对于该属的生态生理学研究很重要。我们使用全基因组序列通过多种分析(16S rRNA、MLSA、氨基酸使用情况、ANI)来评估系统发育关系,这使我们能够识别出两个错误鉴定的菌株。基因组分析还揭示了高GC含量和低GC含量分支的存在,这与系统发育簇相关。泛基因组和核心基因组分析表明,整个属中25%的基因组是保守的,具有一个庞大且开放的泛基因组。基因组多样性的主要来源可追溯到较小的染色体和质粒。该属中研究的几个生理特征与系统发育数据不相关。由于水平基因转移(HGT)常被认为是细菌物种遗传多样性的来源和基因组进化的潜在驱动力,我们研究了该属基因组中此类证据。基因组岛是同一物种菌株之间基因组差异的来源。此外,我们发现了转座酶基因和CRISPR阵列,这表明与外源DNA有多次接触。基因组交换特征在该属中广泛且丰富地存在,表明其在基因组进化中起作用。高遗传变异性和遗传交换迹象使得难以阐明基因组进化路径,并提高了对外源DNA在环境生物基因组进化中作用的认识。