Bonjack-Shterengartz Maayan, Avnir David
Institute of Chemistry and the Lise Meitner Minerva Center for Computational Quantum Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel.
PLoS One. 2017 Jul 14;12(7):e0180030. doi: 10.1371/journal.pone.0180030. eCollection 2017.
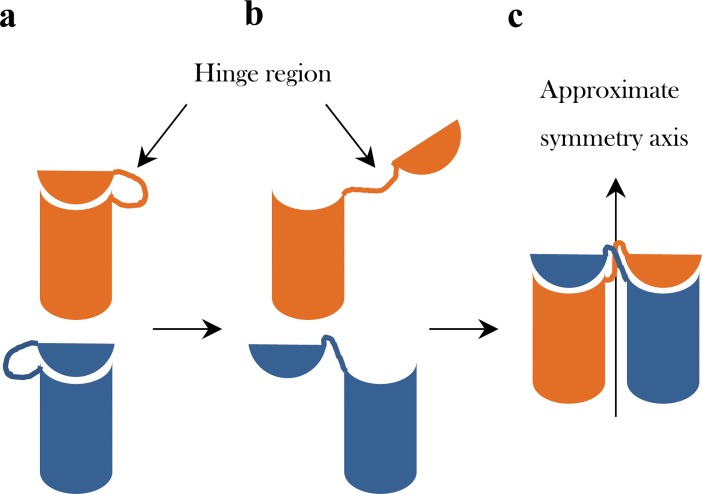
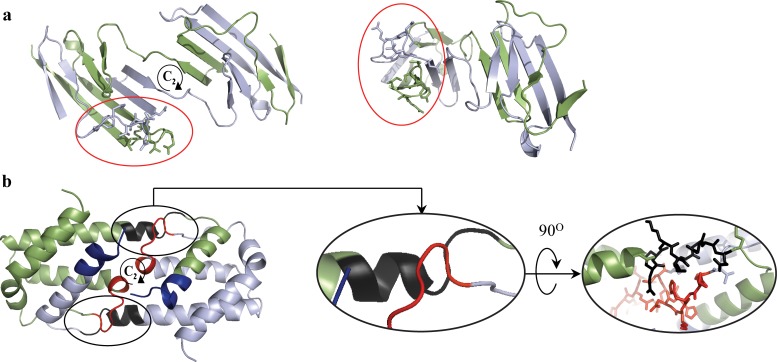
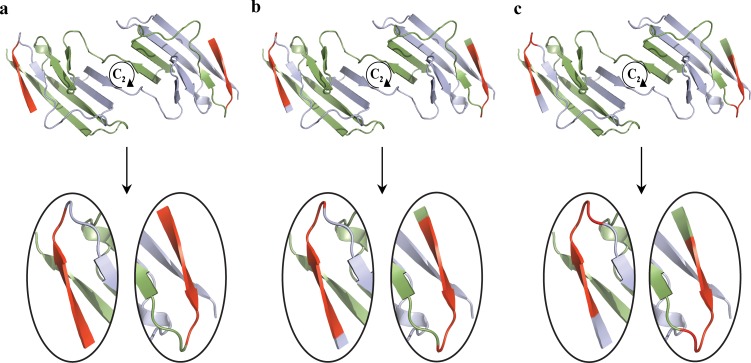
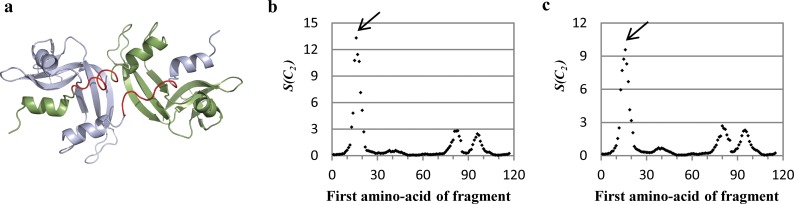
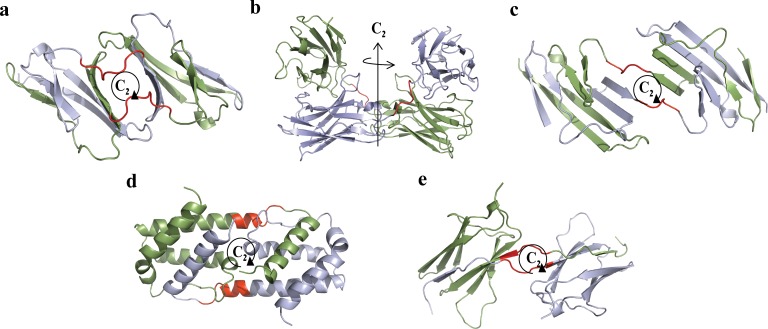
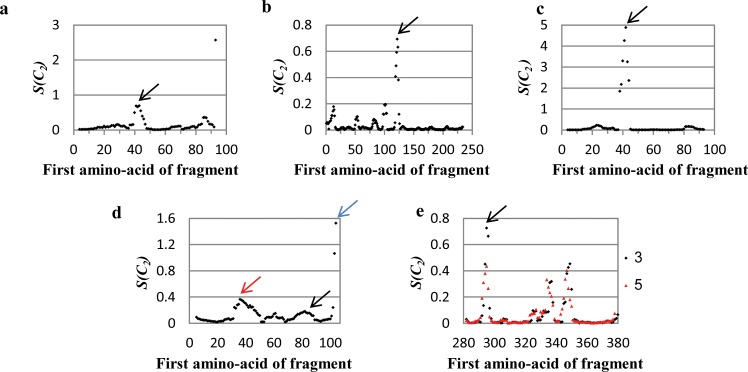
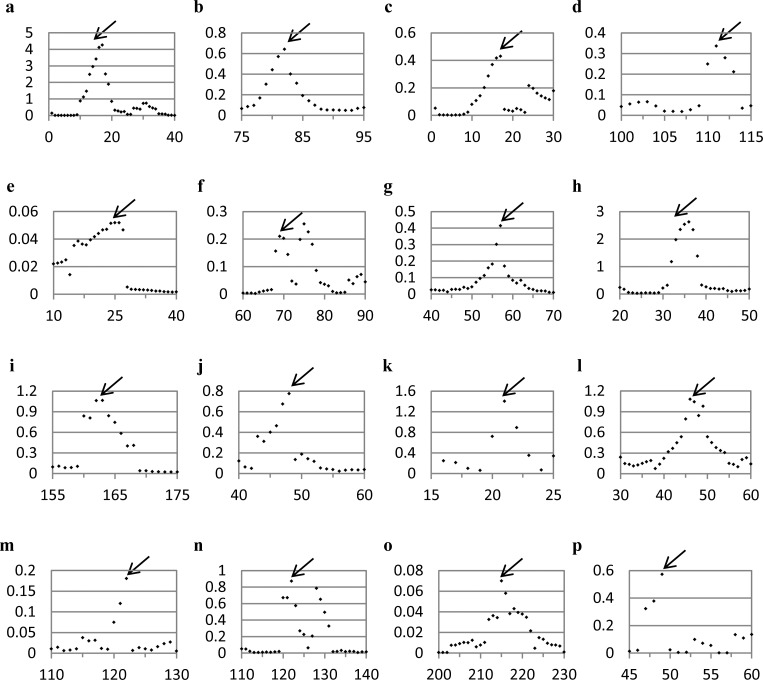
The majority of proteins form oligomers which have rotational symmetry. Literature has suggested many functional advantages that the symmetric packing offers. Yet, despite these advantages, the vast majority of protein oligomers are only nearly symmetric. A key question in the field of proteins structure is therefore, if symmetry is so advantageous, why do oligomers settle for aggregates that do not maximize that structural property? The answer to that question is apparently multi-parametric, and involves distortions at the interaction zones of the monomer units of the oligomer in order to minimize the free energy, the dynamics of the protein, the effects of surroundings parameters, and the mechanism of oligomerization. The study of this problem is in its infancy: Only the first parameter has been explored so far. Here we focus on the last parameter-the mechanism of formation. To test this effect we have selected to focus on the domain swapping mechanism of oligomerization, by which oligomers form in a mechanism that swaps identical portions of monomeric units, resulting in an interwoven oligomer. We are using continuous symmetry measures to analyze in detail the oligomer formed by this mechanism, and found, that without exception, in all analyzed cases, perfect symmetry is given away, and we are able to identify that the main burden of distortion lies in the hinge regions that connect the swapped portions. We show that the continuous symmetry analysis method clearly identifies the hinge region of swapped domain proteins-considered to be a non-trivial task. We corroborate our conclusion about the central role of the hinge region in affecting the symmetry of the oligomers, by a special probability analysis developed particularly for that purpose.
大多数蛋白质形成具有旋转对称性的寡聚体。文献表明对称堆积具有许多功能优势。然而,尽管有这些优势,绝大多数蛋白质寡聚体只是近乎对称。因此,蛋白质结构领域的一个关键问题是,如果对称性如此有利,为什么寡聚体满足于不能使该结构特性最大化的聚集体?这个问题的答案显然是多参数的,并且涉及寡聚体单体单元相互作用区域的扭曲,以最小化自由能、蛋白质的动力学、周围环境参数的影响以及寡聚化机制。对这个问题的研究尚处于起步阶段:到目前为止只探讨了第一个参数。在这里,我们关注最后一个参数——形成机制。为了测试这种效应,我们选择关注寡聚化的结构域交换机制,通过这种机制,寡聚体以交换单体单元相同部分的方式形成,从而产生交织的寡聚体。我们使用连续对称性测量来详细分析通过这种机制形成的寡聚体,并且发现,在所有分析的情况下无一例外,完美对称性都丧失了,而且我们能够确定扭曲的主要负担在于连接交换部分的铰链区域。我们表明连续对称性分析方法能够清晰地识别结构域交换蛋白的铰链区域——这被认为是一项艰巨的任务。我们通过专门为此目的开发的特殊概率分析,证实了我们关于铰链区域在影响寡聚体对称性方面核心作用的结论。