Division of Experimental Chemotherapy, Cancer Chemotherapy Center, Japanese Foundation for Cancer Research, Tokyo, 135-8550, Japan.
Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Tokyo, 108-8639, Japan.
Sci Rep. 2017 Jul 17;7(1):5519. doi: 10.1038/s41598-017-05736-9.
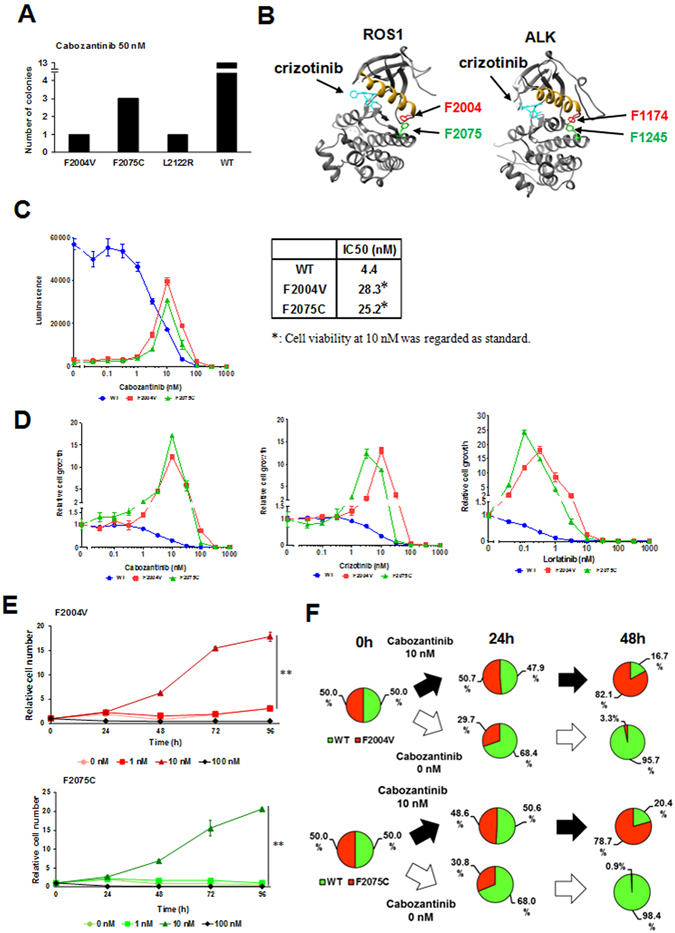
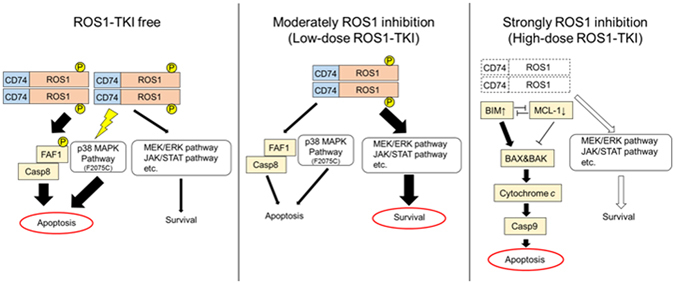
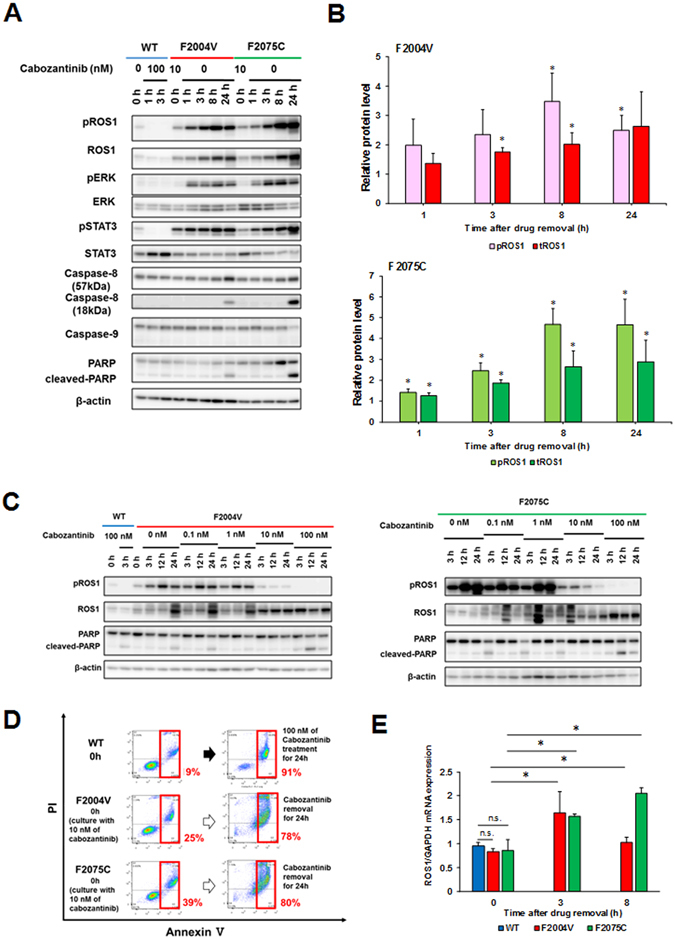
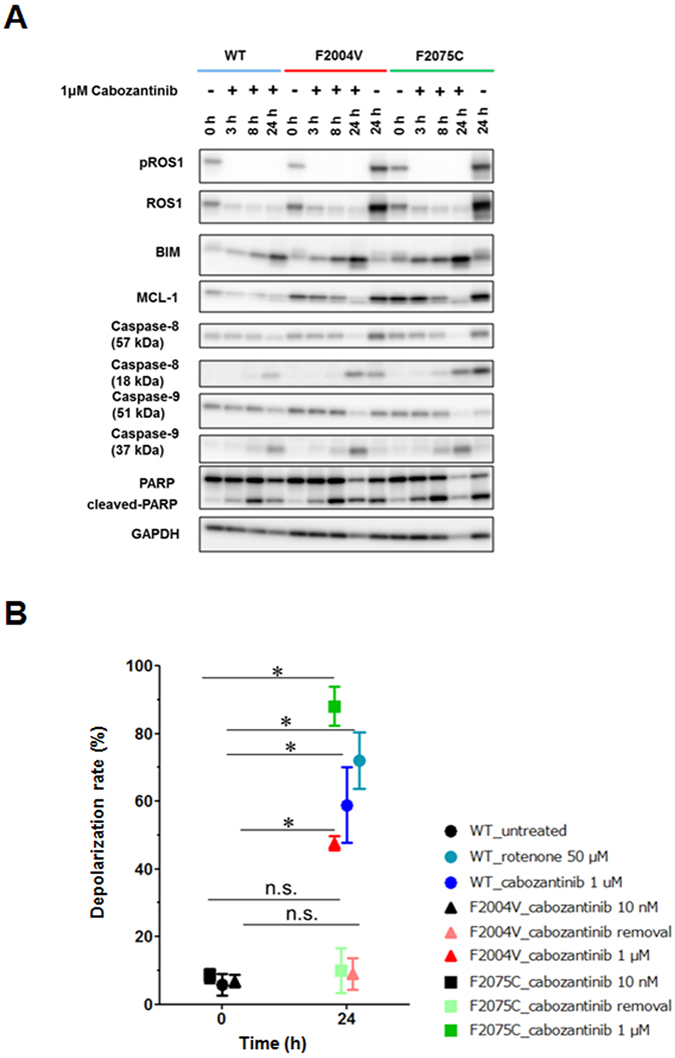
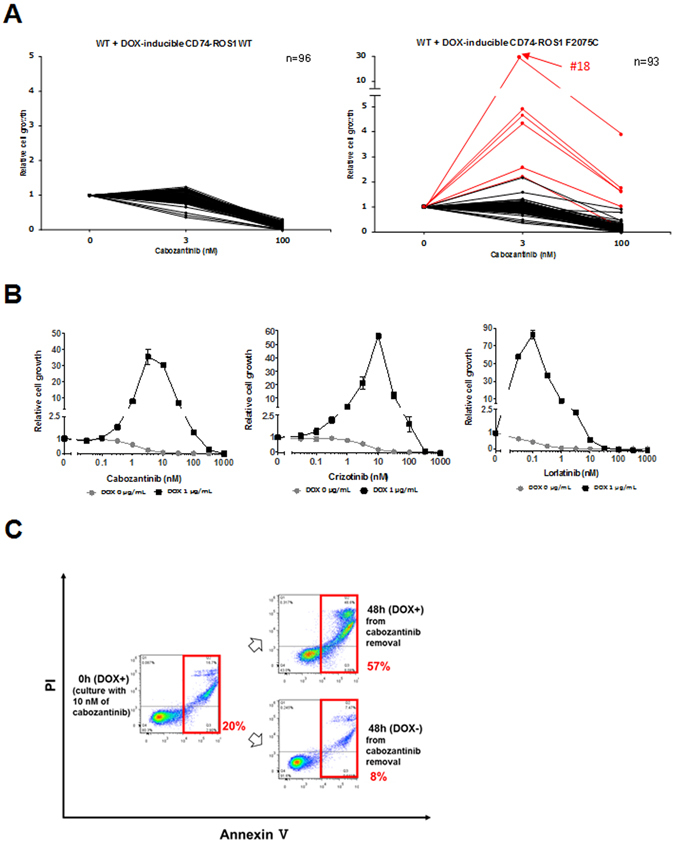
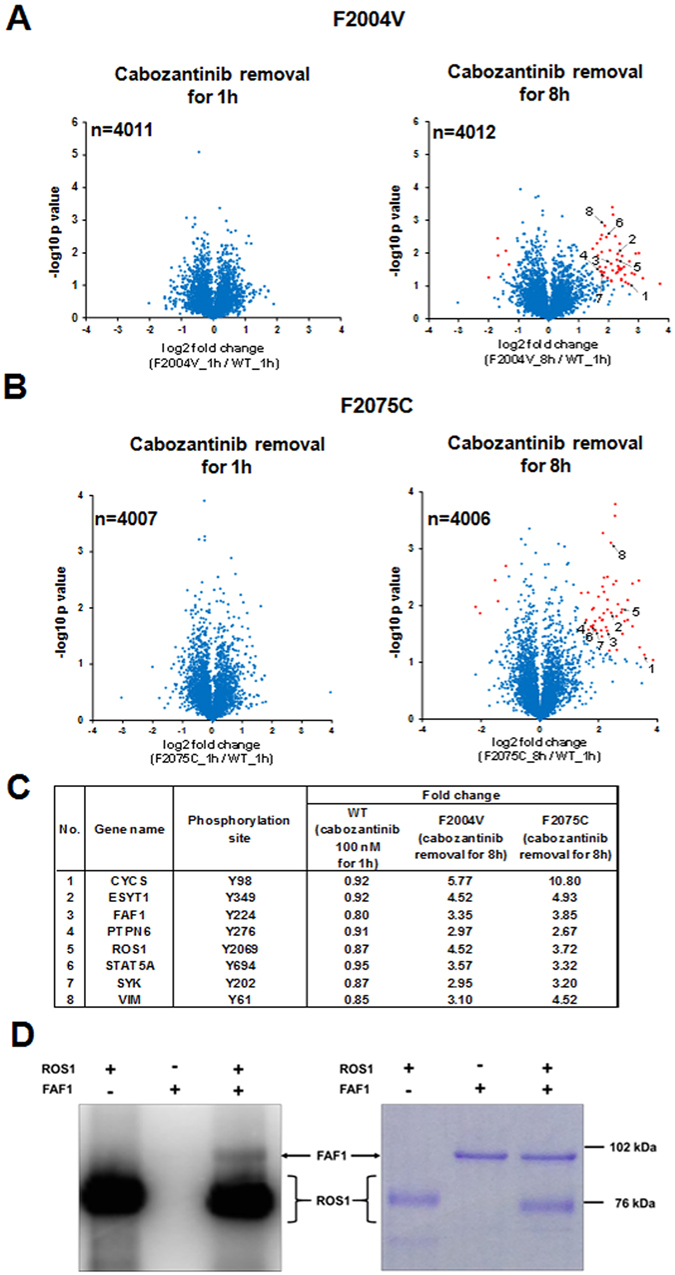
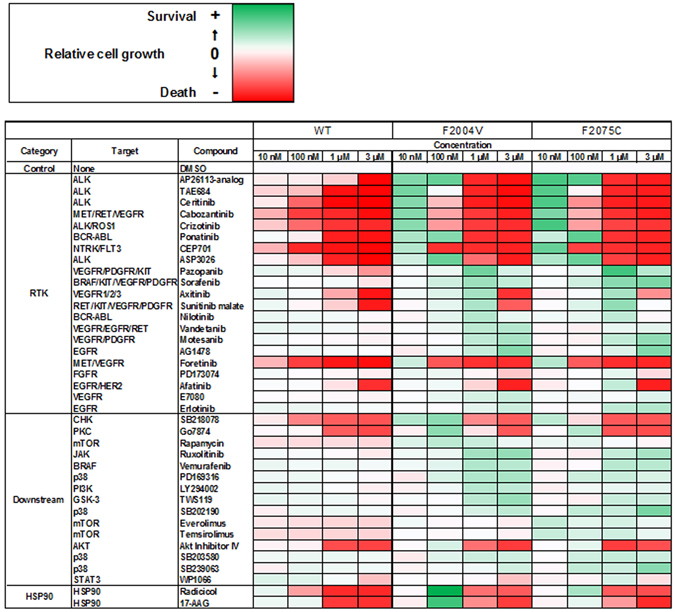
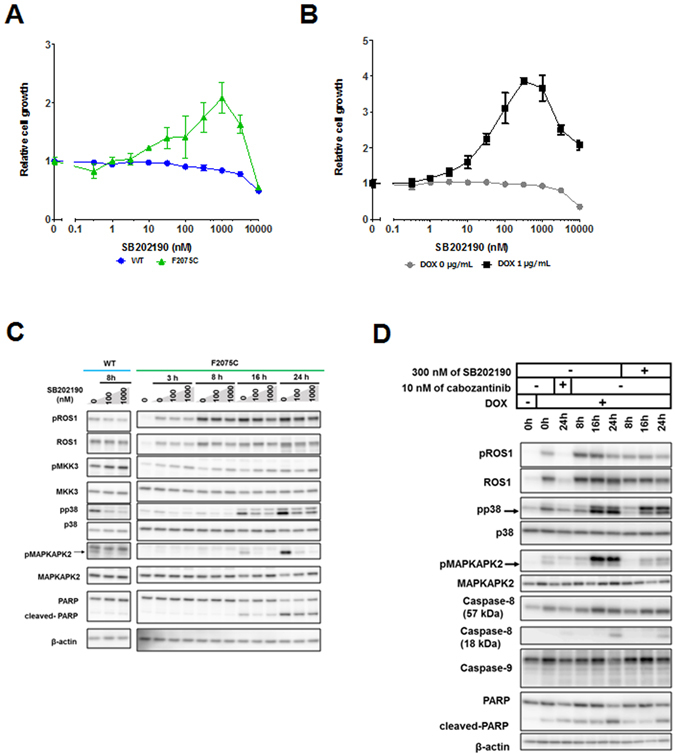
ROS1 rearrangement is observed in 1-2% of non-small cell lung cancers (NSCLC). The ROS1 tyrosine kinase inhibitor (TKI) crizotinib has induced marked tumour shrinkage in ROS1-rearranged cancers. However, emergence of acquired resistance to TKI is inevitable within a few years. Previous findings indicate that cabozantinib overcomes secondary mutation-mediated crizotinib-resistance in ROS1-fusion-positive cells. Here we attempted to establish cabozantinib-resistant cells by N-ethyl-N-nitrosourea mutagenesis screening using CD74-ROS1-expressing Ba/F3 cells. Two resistant cell lines with CD74-ROS1 F2004V or F2075C mutations, which are homologous to ALK F1174 or F1245 mutations, survived in the presence of a low dose of ROS1-TKI. Removal of ROS1-TKI from these TKI-addicted cells induced excessive activation of ROS1 tyrosine kinase followed by apoptosis. We succeeded in recapturing the TKI-addicted phenotype using doxycycline-inducible CD74-ROS1 mutant over-expression in Ba/F3 cells, suggesting that excessive ROS1 oncogenic signaling itself induced apoptosis instead of cell growth. Phosphoproteomic analysis and high-throughput inhibitor screening revealed that excessive ROS1 signaling in the TKI-addicted cells phosphorylated or activated apoptosis-related molecules such as FAF1 or p38. Collectively, our findings partly clarify molecular mechanisms of excessive ROS1 oncogenic signaling that mediates paradoxical induction of apoptosis.
ROS1 重排发生于 1%-2%的非小细胞肺癌(NSCLC)中。ROS1 酪氨酸激酶抑制剂(TKI)克唑替尼已使 ROS1 重排肿瘤明显缩小。然而,TKI 获得性耐药在数年内不可避免。先前的研究结果表明卡博替尼克服了 ROS1 融合阳性细胞中继发性突变介导的克唑替尼耐药。本研究通过 N-乙基-N-亚硝基脲诱变筛选,在表达 CD74-ROS1 的 Ba/F3 细胞中尝试建立卡博替尼耐药细胞系。在低剂量 ROS1-TKI 存在下存活的两种耐药细胞系具有 CD74-ROS1 F2004V 或 F2075C 突变,与 ALK F1174 或 F1245 突变同源。在这些 TKI 成瘾细胞中去除 ROS1-TKI 会导致 ROS1 酪氨酸激酶过度激活,随后发生细胞凋亡。我们成功地通过在 Ba/F3 细胞中使用强力霉素诱导型 CD74-ROS1 突变体过表达重新捕获 TKI 成瘾表型,这表明ROS1 致癌信号本身的过度激活诱导细胞凋亡而不是细胞生长。磷酸化蛋白质组学分析和高通量抑制剂筛选显示,TKI 成瘾细胞中过度的 ROS1 信号转导磷酸化或激活了 FAF1 或 p38 等与凋亡相关的分子。总之,我们的研究结果部分阐明了介导矛盾性凋亡诱导的过度 ROS1 致癌信号转导的分子机制。