Bholah Reshma, Bunchman Timothy Edward
Pediatric Nephrology, Virginia Commonwealth University, Richmond, VA, United States.
Front Pediatr. 2017 Jul 13;5:155. doi: 10.3389/fped.2017.00155. eCollection 2017.
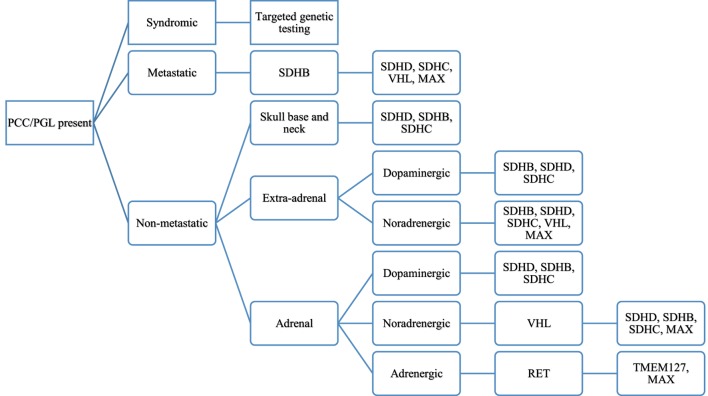
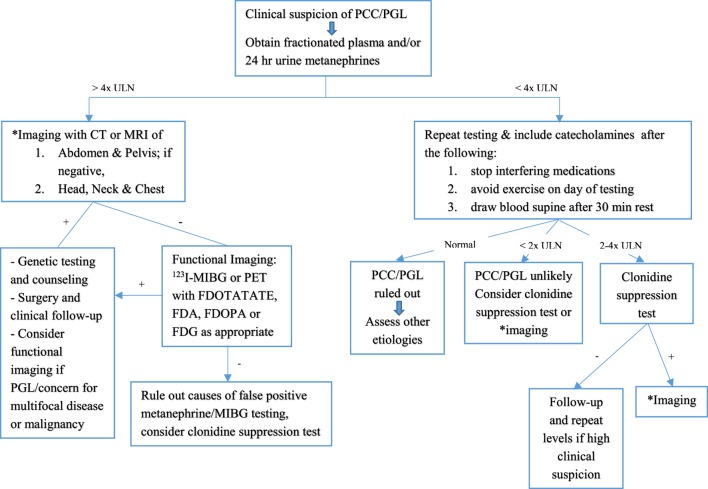
Pheochromocytoma (PCC) and paraganglioma (PGL) are rare chromaffin cell tumors which secrete catecholamines and form part of the family of neuroendocrine tumors. Although a rare cause of secondary hypertension in pediatrics, the presentation of hypertension in these patients is characteristic, and treatment is definitive. The gold standard for diagnosis is measurement of plasma free metanephrines, with imaging studies performed for localization, identification of metastatic lesions and for surgical resection. Preoperative therapy with alpha-blocking agents, beta blockers, and potentially tyrosine hydroxylase inhibitors aid in a safe pre-, intra- and postoperative course. PCC and PGL are inherited in as much as 80% of pediatric cases, and all patients with mutations should be followed closely given the risk of recurrence and malignancy. While the presentation of chromaffin cell tumors has been well described with multiple endocrine neoplasia, NF1, and Von Hippel-Lindau syndromes, the identification of new gene mutations leading to chromaffin cell tumors at a young age is changing the landscape of how clinicians approach such cases. The paraganglioma-pheochromocytoma syndromes (SDHx) comprise familial gene mutations, of which the SDHB gene mutation carries a high rate of malignancy. Since the inheritance rate of such tumors is higher than previously described, genetic screening is recommended in all patients, and lifelong follow-up for recurrent tumors is a must. A multidisciplinary team approach allows for optimal health-care delivery in such children. This review serves to provide an overview of pediatric PCC and PGL, including updates on the preferred methods of imaging, guidelines on gene testing as well as management of hypertension in such patients.
嗜铬细胞瘤(PCC)和副神经节瘤(PGL)是罕见的嗜铬细胞瘤,可分泌儿茶酚胺,属于神经内分泌肿瘤家族的一部分。虽然是小儿继发性高血压的罕见病因,但这些患者的高血压表现具有特征性,且治疗具有确定性。诊断的金标准是测定血浆游离甲氧基肾上腺素,并进行影像学检查以定位、识别转移灶和进行手术切除。术前使用α受体阻滞剂、β受体阻滞剂以及可能的酪氨酸羟化酶抑制剂有助于实现安全的术前、术中和术后过程。在小儿病例中,多达80%的PCC和PGL是遗传性的,鉴于复发和恶性风险,所有有突变的患者都应密切随访。虽然嗜铬细胞瘤的表现已在多发性内分泌肿瘤、1型神经纤维瘤病(NF1)和冯希佩尔-林道综合征中得到充分描述,但在年轻时发现导致嗜铬细胞瘤的新基因突变正在改变临床医生处理此类病例的方式。副神经节瘤-嗜铬细胞瘤综合征(SDHx)包括家族性基因突变,其中SDHB基因突变的恶性率很高。由于此类肿瘤的遗传率高于先前描述的,建议对所有患者进行基因筛查,并且对复发性肿瘤进行终身随访是必需的。多学科团队方法可为此类儿童提供最佳医疗服务。本综述旨在概述小儿PCC和PGL,包括影像学首选方法的更新、基因检测指南以及此类患者高血压的管理。