Aziz Md Momin Al, Alhadidi Dima, Mohammed Noman
Department of Computer Science, University of Manitoba, Winnipeg, Canada.
Zayed University, Dubai, United Arab Emirates.
BMC Med Genomics. 2017 Jul 26;10(Suppl 2):41. doi: 10.1186/s12920-017-0279-9.
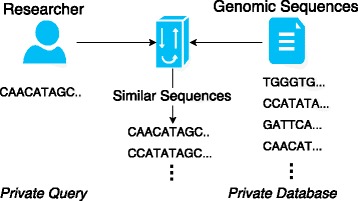
Edit distance is a well established metric to quantify how dissimilar two strings are by counting the minimum number of operations required to transform one string into the other. It is utilized in the domain of human genomic sequence similarity as it captures the requirements and leads to a better diagnosis of diseases. However, in addition to the computational complexity due to the large genomic sequence length, the privacy of these sequences are highly important. As these genomic sequences are unique and can identify an individual, these cannot be shared in a plaintext.
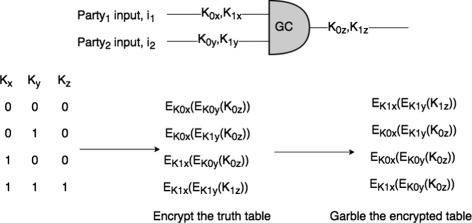
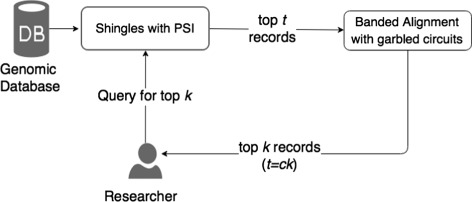
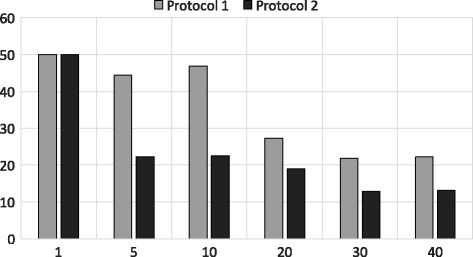
In this paper, we propose two different approximation methods to securely compute the edit distance among genomic sequences. We use shingling, private set intersection methods, the banded alignment algorithm, and garbled circuits to implement these methods. We experimentally evaluate these methods and discuss both advantages and limitations.
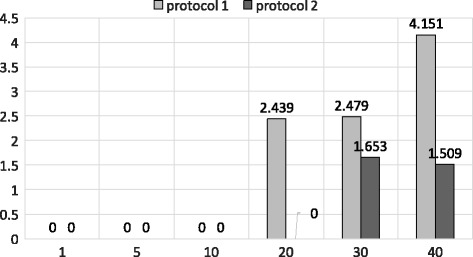
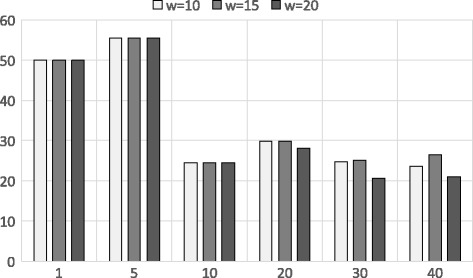
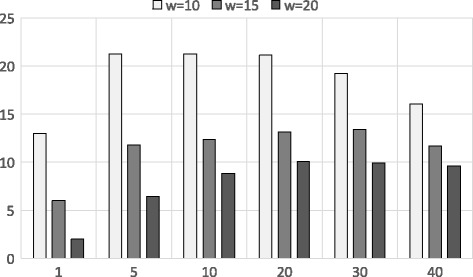
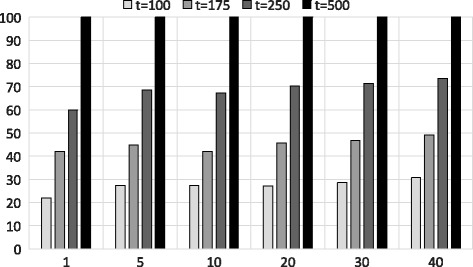
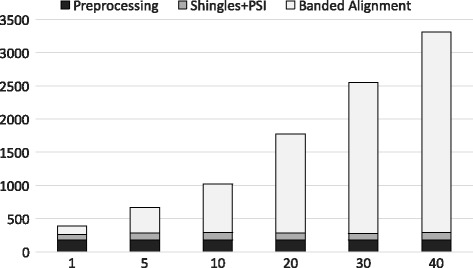
Experimental results show that our first approximation method is fast and achieves similar accuracy compared to existing techniques. However, for longer genomic sequences, both the existing techniques and our proposed first method are unable to achieve a good accuracy. On the other hand, our second approximation method is able to achieve higher accuracy on such datasets. However, the second method is relatively slower than the first proposed method.
The proposed algorithms are generally accurate, time-efficient and can be applied individually and jointly as they have complimentary properties (runtime vs. accuracy) on different types of datasets.
编辑距离是一种成熟的度量标准,通过计算将一个字符串转换为另一个字符串所需的最少操作数来量化两个字符串的差异程度。它被应用于人类基因组序列相似性领域,因为它能够满足需求并有助于更好地诊断疾病。然而,除了由于基因组序列长度大导致的计算复杂性外,这些序列的隐私性也非常重要。由于这些基因组序列是唯一的且能够识别个体,所以不能以明文形式共享。
在本文中,我们提出了两种不同的近似方法来安全地计算基因组序列之间的编辑距离。我们使用分片、私有集合交集方法、带状比对算法和混淆电路来实现这些方法。我们通过实验对这些方法进行评估,并讨论其优缺点。
实验结果表明,我们的第一种近似方法速度快,与现有技术相比具有相似的准确性。然而,对于较长的基因组序列,现有技术和我们提出的第一种方法都无法达到良好的准确性。另一方面,我们的第二种近似方法在此类数据集上能够达到更高的准确性。然而,第二种方法比第一种方法相对较慢。
所提出的算法总体上准确、高效,并且由于它们在不同类型的数据集上具有互补特性(运行时间与准确性),可以单独应用或联合应用。