Lipska-Ziętkiewicz Beata S, Gellermann Jutta, Boyer Olivia, Gribouval Olivier, Ziętkiewicz Szymon, Kari Jameela A, Shalaby Mohamed A, Ozaltin Fatih, Dusek Jiri, Melk Anette, Bayazit Aysun K, Massella Laura, Hyla-Klekot Lidia, Habbig Sandra, Godron Astrid, Szczepańska Maria, Bieniaś Beata, Drożdż Dorota, Odeh Rasha, Jarmużek Wioletta, Zachwieja Katarzyna, Trautmann Agnes, Antignac Corinne, Schaefer Franz
Department of Biology and Medical Genetics, Clinical Genetics Unit, Medical University of Gdansk, Gdansk, Poland.
Department of Pediatric Nephrology, Charité Universitätsmedizin Berlin, Charité Children's Hospital, Berlin, Germany.
PLoS One. 2017 Aug 10;12(8):e0180926. doi: 10.1371/journal.pone.0180926. eCollection 2017.
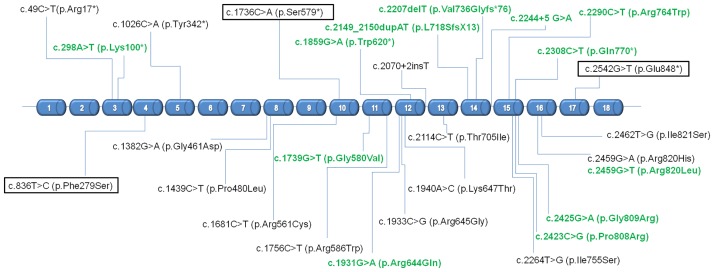
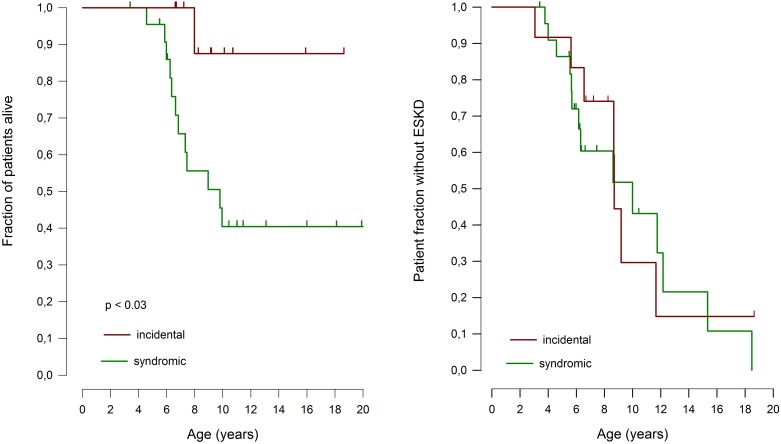
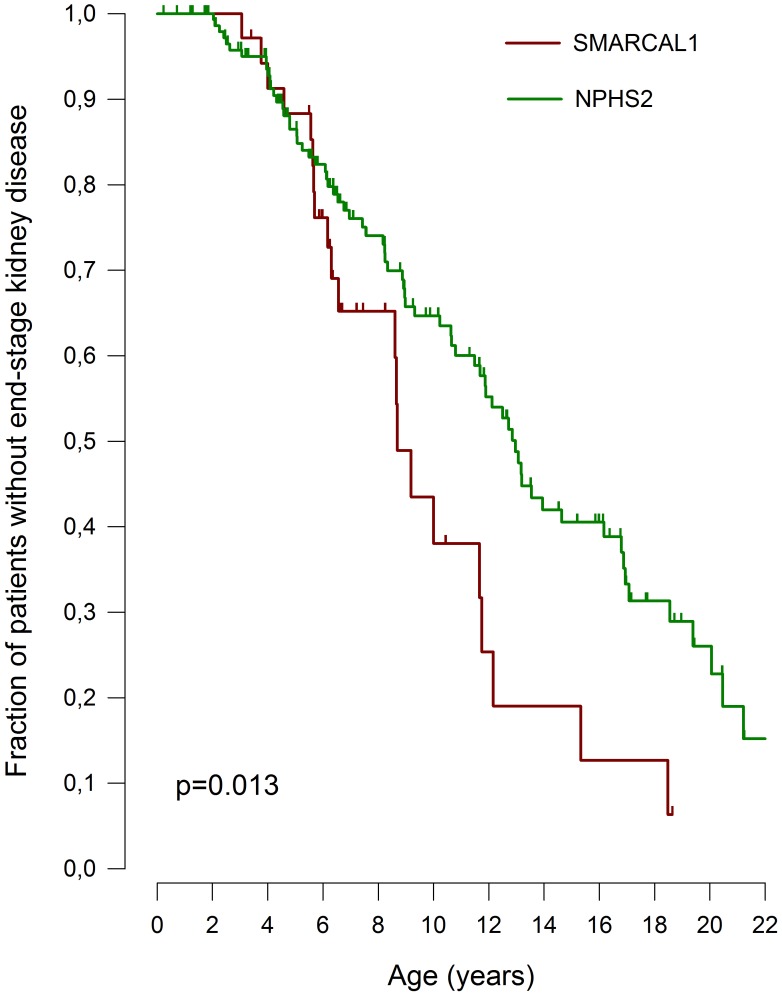
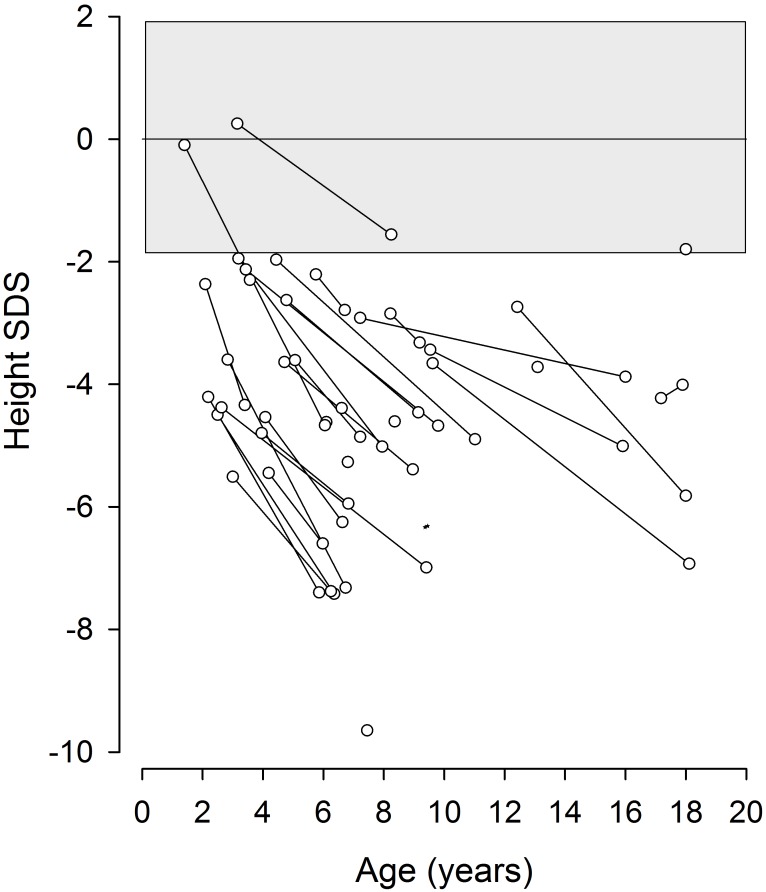
Schimke immuno-osseous dysplasia (SIOD) is a rare multisystem disorder with early mortality and steroid-resistant nephrotic syndrome (SRNS) progressing to end-stage kidney disease. We hypothesized that next-generation gene panel sequencing may unsurface oligosymptomatic cases of SIOD with potentially milder disease courses. We analyzed the renal and extrarenal phenotypic spectrum and genotype-phenotype associations in 34 patients from 28 families, the largest SMARCAL1-associated nephropathy cohort to date. In 11 patients the diagnosis was made unsuspectedly through SRNS gene panel testing. Renal disease first manifested at median age 4.5 yrs, with focal segmental glmerulosclerosis or minimal change nephropathy on biopsy and rapid progression to end-stage kidney disease (ESKD) at median age 8.7 yrs. Whereas patients diagnosed by phenotype more frequently developed severe extrarenal complications (cerebral ischemic events, septicemia) and were more likely to die before age 10 years than patients identified by SRNS-gene panel screening (88 vs. 40%), the subgroups did not differ with respect to age at proteinuria onset and progression to ESKD. Also, 10 of 11 children diagnosed unsuspectedly by Next Generation Sequencing were small at diagnosis and all showed progressive growth failure. Severe phenotypes were usually associated with biallelic truncating mutations and milder phenotypes with biallelic missense mutations. However, no genotype-phenotype correlation was observed for the renal disease course. In conclusion, while short stature is a reliable clue to SIOD in children with SRNS, other systemic features are highly variable. Our findings support routine SMARCAL1 testing also in non-syndromic SRNS.
施姆克免疫性骨发育不良(SIOD)是一种罕见的多系统疾病,具有早期死亡率,且类固醇抵抗性肾病综合征(SRNS)会进展为终末期肾病。我们推测,新一代基因检测板测序可能会发现症状轻微、疾病进程可能较为缓和的SIOD病例。我们分析了来自28个家庭的34例患者的肾脏和肾外表型谱以及基因型-表型关联,这是迄今为止最大的与SMARCAL1相关的肾病队列。在11例患者中,通过SRNS基因检测板检测意外做出了诊断。肾病首次出现的中位年龄为4.5岁,活检显示为局灶节段性肾小球硬化或微小病变性肾病,并在中位年龄8.7岁时迅速进展为终末期肾病(ESKD)。与通过SRNS基因检测板筛查确诊的患者相比,通过表型诊断的患者更常出现严重的肾外并发症(脑缺血事件、败血症),且在10岁前死亡的可能性更大(88%对40%),但这两个亚组在蛋白尿出现年龄和进展至ESKD方面并无差异。此外,11例通过下一代测序意外确诊的儿童中有10例在诊断时身材矮小,且均表现出进行性生长发育迟缓。严重表型通常与双等位基因截短突变相关,较轻表型与双等位基因错义突变相关。然而,未观察到肾病病程与基因型-表型之间的相关性。总之,虽然身材矮小是SRNS患儿SIOD的可靠线索,但其他全身特征差异很大。我们的研究结果支持在非综合征性SRNS中也进行常规的SMARCAL1检测。