Nascimento Clarissa R, Andrade Daniele, Carvalho-Pinto Carla Eponina, Serra Rafaela Rangel, Vellasco Lucas, Brasil Guilherme, Ramos-Junior Erivan Schnaider, da Mota Julia Barbalho, Almeida Larissa Nogueira, Andrade Marcus V, Correia Soeiro Maria de Nazaré, Juliano Luiz, Alvarenga Patrícia Hessab, Oliveira Ana Carolina, Sicuro Fernando Lencastre, de Carvalho Antônio C Campos, Svensjö Erik, Scharfstein Julio
Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil.
Departamento de Imunobiologia, Universidade Federal Fluminense (UFF), Niterói, Brazil.
Front Immunol. 2017 Aug 2;8:840. doi: 10.3389/fimmu.2017.00840. eCollection 2017.
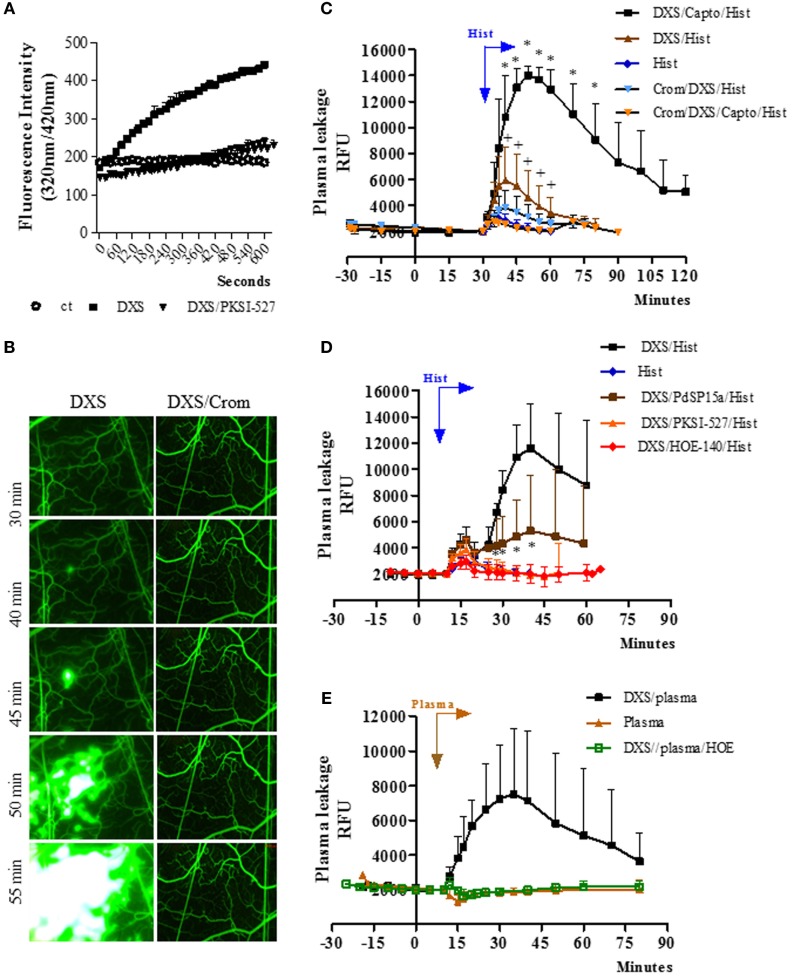
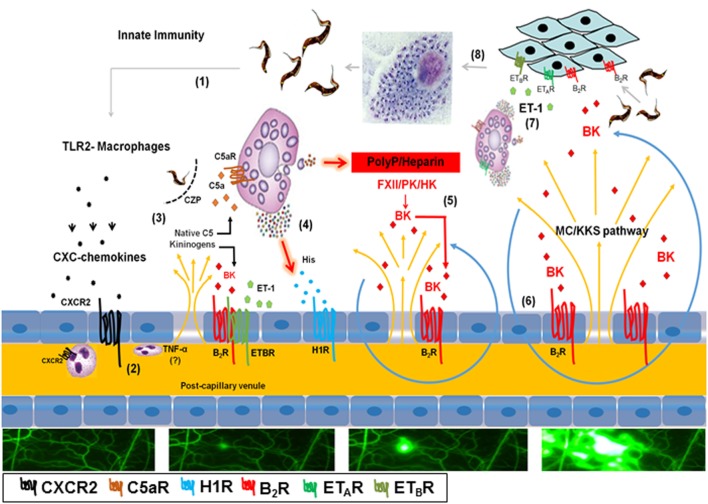
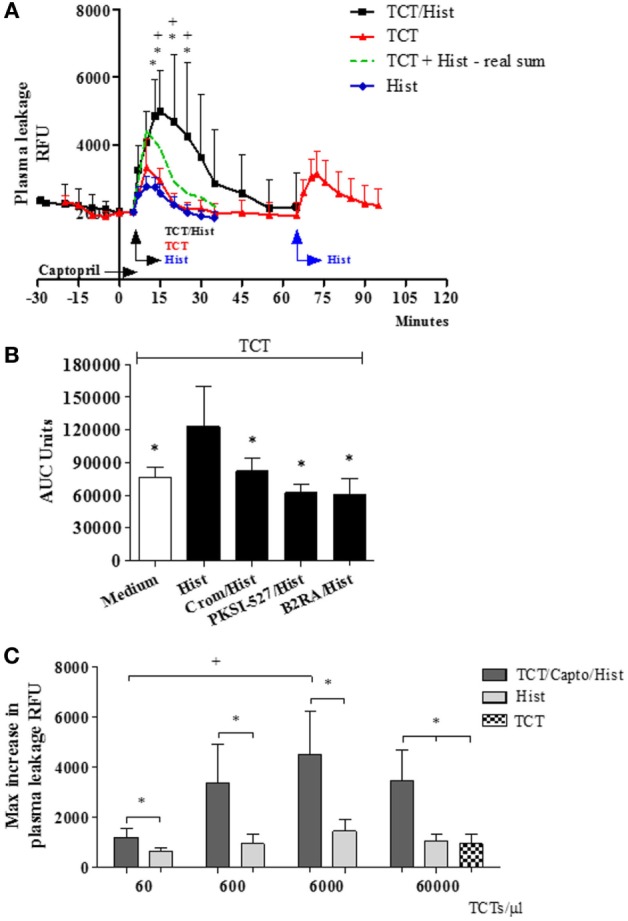
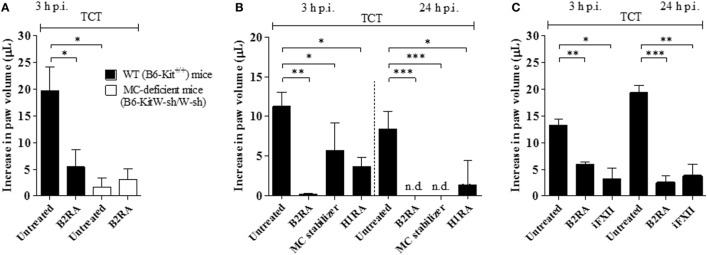
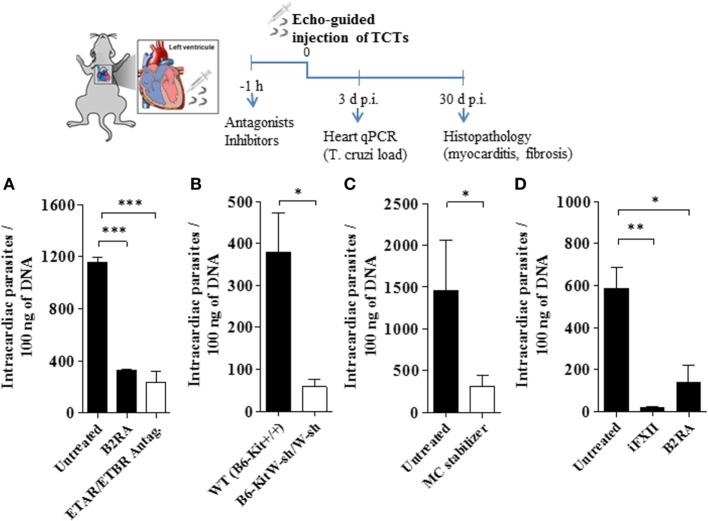
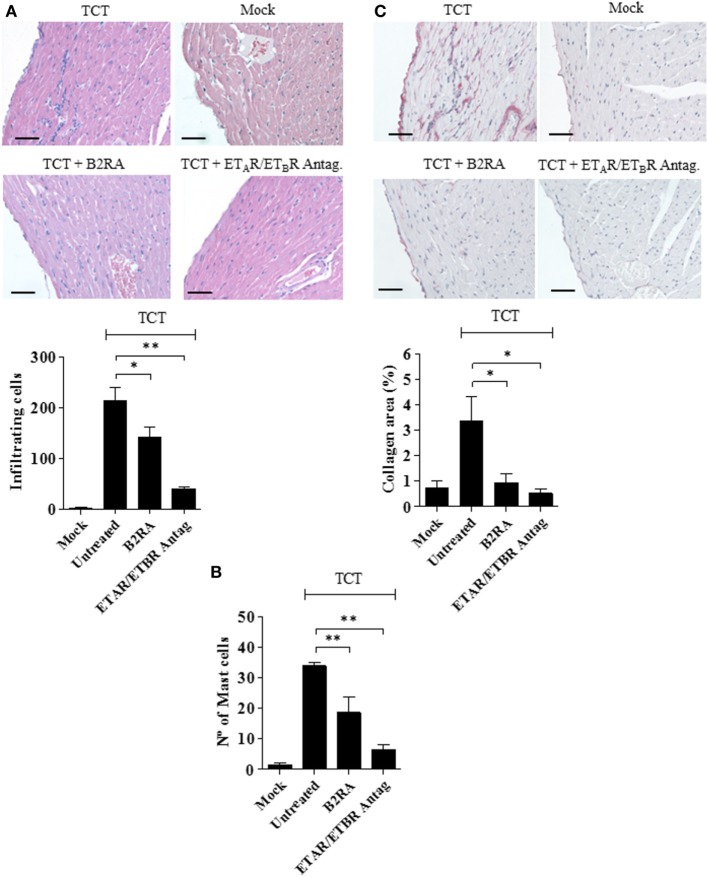
During the course of Chagas disease, infectious forms of are occasionally liberated from parasitized heart cells. Studies performed with tissue culture trypomastigotes (TCTs, Dm28c strain) demonstrated that these parasites evoke neutrophil/CXCR2-dependent microvascular leakage by activating innate sentinel cells toll-like receptor 2 (TLR2). Upon plasma extravasation, proteolytically derived kinins and C5a stimulate immunoprotective Th1 responses cross-talk between bradykinin B2 receptors (B2Rs) and C5aR. Awareness that TCTs invade cardiovascular cells interdependent activation of B2R and endothelin receptors [endothelin A receptor (ETR)/endothelin B receptor (ETR)] led us to hypothesize that might reciprocally benefit from the formation of infection-associated edema activation of kallikrein-kinin system (KKS). Using intravital microscopy, here we first examined the functional interplay between mast cells (MCs) and the KKS by topically exposing the hamster cheek pouch (HCP) tissues to dextran sulfate (DXS), a potent "contact" activator of the KKS. Surprisingly, although DXS was inert for at least 30 min, a subtle MC-driven leakage resulted in factor XII (FXII)-dependent activation of the KKS, which then amplified inflammation generation of bradykinin (BK). Guided by this mechanistic insight, we next exposed TCTs to "leaky" HCP-forged by low dose histamine application-and found that the proinflammatory phenotype of TCTs was boosted by BK generated the MC/KKS pathway. Measurements of footpad edema in MC-deficient mice linked TCT-evoked inflammation to MC degranulation (upstream) and FXII-mediated generation of BK (downstream). We then inoculated TCTs intracardiacally in mice and found a striking decrease of parasite DNA (quantitative polymerase chain reaction; 3 d.p.i.) in the heart of MC-deficient mutant mice. Moreover, the intracardiac parasite load was significantly reduced in WT mice pretreated with (i) cromoglycate (MC stabilizer) (ii) infestin-4, a specific inhibitor of FXIIa (iii) HOE-140 (specific antagonist of B2R), and (iv) bosentan, a non-selective antagonist of ETR/ETR. Notably, histopathology of heart tissues from mice pretreated with these G protein-coupled receptors blockers revealed that myocarditis and heart fibrosis (30 d.p.i.) was markedly and redundantly attenuated. Collectively, our study suggests that inflammatory edema propagated activation of the MC/KKS pathway fuels intracardiac parasitism by generating infection-stimulatory peptides (BK and endothelins) in the edematous heart tissues.
在恰加斯病病程中,感染性形式的[此处原文缺失具体内容]偶尔会从被寄生的心脏细胞中释放出来。对组织培养型锥鞭毛体(TCTs,Dm28c株)进行的研究表明,这些寄生虫通过激活天然哨兵细胞 toll样受体2(TLR2)引发中性粒细胞/CXCR2依赖性微血管渗漏。血浆外渗时,蛋白水解衍生的激肽和C5a刺激免疫保护性Th1反应——缓激肽B2受体(B2Rs)和C5aR之间的相互作用。意识到TCTs侵入心血管细胞——B2R和内皮素受体[内皮素A受体(ETR)/内皮素B受体(ETR)]的相互依赖性激活——使我们推测[此处原文缺失具体内容]可能从感染相关水肿的形成——激肽释放酶-激肽系统(KKS)的激活——中相互受益。在这里,我们使用活体显微镜,首先通过将仓鼠颊囊(HCP)组织局部暴露于硫酸葡聚糖(DXS)(一种有效的KKS“接触”激活剂)来研究肥大细胞(MCs)与KKS之间的功能相互作用。令人惊讶的是,尽管DXS至少30分钟内无活性,但微妙的MC驱动渗漏导致了因子XII(FXII)依赖性的KKS激活,随后炎症放大并产生缓激肽(BK)。基于这一机制性见解,我们接下来将TCTs暴露于通过低剂量组胺应用形成的“渗漏性”HCP中,发现TCTs的促炎表型因MC/KKS途径产生的BK而增强。在MC缺陷小鼠中测量足垫水肿,将TCTs诱发的炎症与MC脱颗粒(上游)和FXII介导的BK生成(下游)联系起来。然后我们将TCTs心脏内接种到小鼠体内,发现MC缺陷突变小鼠心脏中的寄生虫DNA(定量聚合酶链反应;感染后3天)显著减少。此外,用(i)色甘酸(MC稳定剂)、(ii)抑肽素-4(FXIIa的特异性抑制剂)、(iii)HOE-140(B2R的特异性拮抗剂)和(iv)波生坦(ETR/ETR的非选择性拮抗剂)预处理的野生型小鼠心脏内寄生虫负荷显著降低。值得注意的是,用这些G蛋白偶联受体阻滞剂预处理的小鼠心脏组织的组织病理学显示,心肌炎和心脏纤维化(感染后30天)明显且多重减轻。总体而言,我们的研究表明,由MC/KKS途径激活传播的炎症性水肿通过在水肿的心脏组织中产生感染刺激肽(BK和内皮素)来促进心脏内寄生。