Department of Regulatory Science, Hokkaido University Graduate School of Medicine, Sapporo, Japan.
Department of Clinical Medicine (Pharmaceutical Medicine), Graduate School of Pharmaceutical Science, Kitasato University, Tokyo, Japan.
Orphanet J Rare Dis. 2017 Aug 23;12(1):143. doi: 10.1186/s13023-017-0690-5.
The unmet medical needs of individuals with very rare diseases are high. The clinical trial designs and evaluation methods used for 'regular' drugs are not applicable in the clinical development of ultra-orphan drugs (<1000 patients) in many cases. In order to improve the clinical development of ultra-orphan drugs, we examined several points regarding the efficient evaluations of drug efficacy and safety that could be conducted even with very small sample sizes, based on the review reports of orphan drugs approved in Japan.
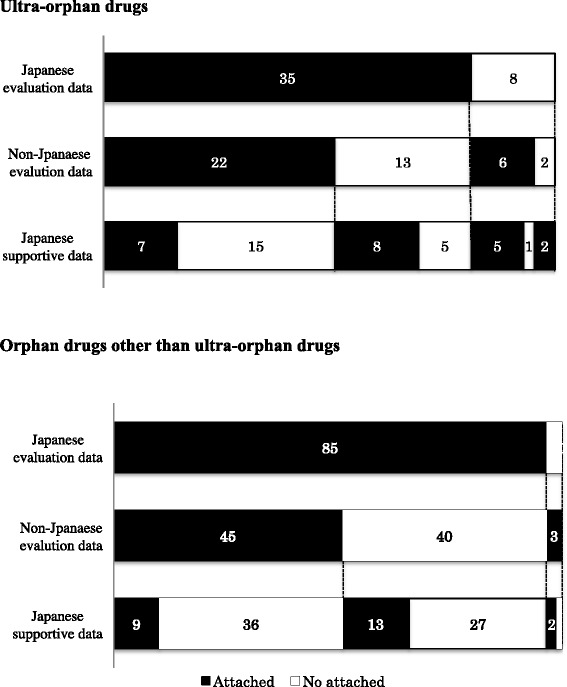
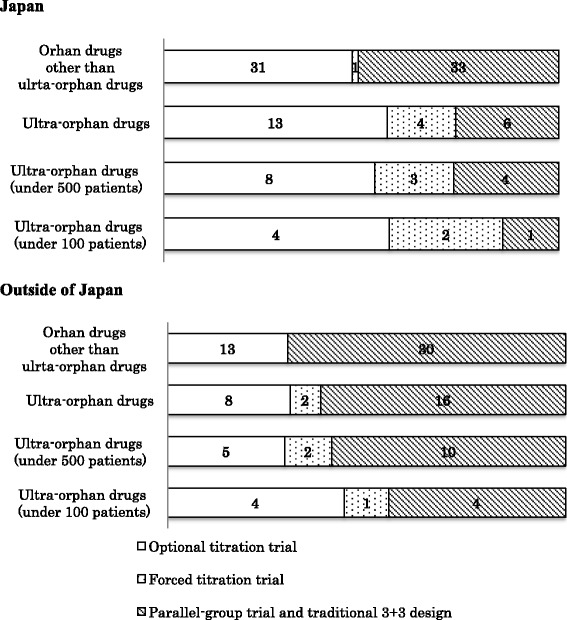
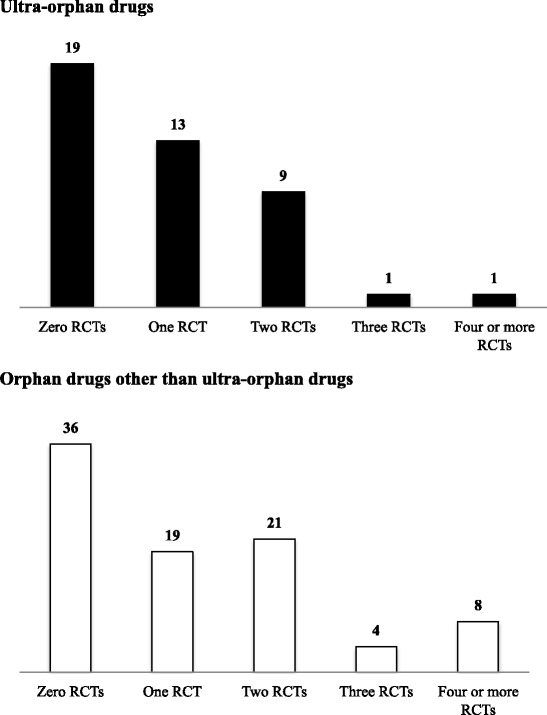
The clinical data packages of 43 ultra-orphan drugs approved in Japan from January 2001 to December 2014 were investigated. Japanese clinical trial data were not included in the clinical data package for eight ultra-orphan drugs, and non-Japanese clinical trial data were included for six of these eight drug. Japanese supportive data that included retrospective studies, published literature, clinical research and Japanese survey results were clinical data package attachments in 22 of the 43 ultra-orphan drugs. Multinational trials were conducted for three ultra-orphan drugs. More than two randomized controlled trials (RCTs) were conducted for only 11 of the 43 ultra-orphan drugs. The smaller the number of patients, the greater the proportion of forced titration and optional titration trials were conducted. Extension trials were carried out for enzyme preparations and monoclonal antibodies with high ratio. Post-marketing surveillance of all patients was required in 36 of the 43 ultra-orphan drugs. For ultra-orphan drugs, clinical endpoints were used as the primary efficacy endpoint of the pivotal trial only for two drugs. The control groups in RCTs were classified as follows: placebo groups different dosage groups, and active controls groups. Sample sizes have been determined on the basis of feasibility for some ultra-orphan drugs. We provide "Draft Guidance on the Clinical Development of Ultra-Orphan Drugs" based on this research.
The development of ultra-orphan drugs requires various arrangements regarding evidence collection, data sources and the clinical trial design. We expect that this draft guidance is useful for ultra-orphan drugs developments in future.
患有极罕见疾病的个体存在着大量未满足的医疗需求。在许多情况下,针对临床开发超罕见药物(<1000 名患者),“常规”药物的临床试验设计和评估方法并不适用。为了提高超罕见药物的临床开发水平,我们根据日本批准的孤儿药审查报告,研究了即使在样本量非常小的情况下,也可以进行的药物疗效和安全性的高效评估的几个要点。
对 2001 年 1 月至 2014 年 12 月期间在日本批准的 43 种超罕见药物的临床数据包进行了调查。8 种超罕见药物的临床数据包中未包含日本临床试验数据,其中 6 种药物包含非日本临床试验数据。22 种超罕见药物的临床数据包中有日本支持性数据,包括回顾性研究、已发表文献、临床研究和日本调查结果。3 种超罕见药物进行了多国试验。只有 11 种超罕见药物进行了超过 2 次随机对照试验(RCT)。患者人数越少,进行强制滴定和可选滴定试验的比例越高。酶制剂和单克隆抗体进行了扩展试验,比例较高。36 种超罕见药物需要对所有患者进行上市后监测。对于超罕见药物,只有 2 种药物将临床终点作为关键试验的主要疗效终点。RCT 中的对照组分为:安慰剂组、不同剂量组和阳性对照组。一些超罕见药物的样本量是基于可行性确定的。我们根据这项研究提供了“超罕见药物临床开发指南草案”。
超罕见药物的开发需要在证据收集、数据源和临床试验设计方面进行各种安排。我们期望本指南草案对未来的超罕见药物开发有所帮助。