Department of Toxicology, University Medical Center Mainz, Mainz, Germany.
Department of Cell and Molecular Biology, Karolinska Institute, Stockholm, Sweden.
Cell Death Dis. 2017 Aug 24;8(8):e3019. doi: 10.1038/cddis.2017.418.
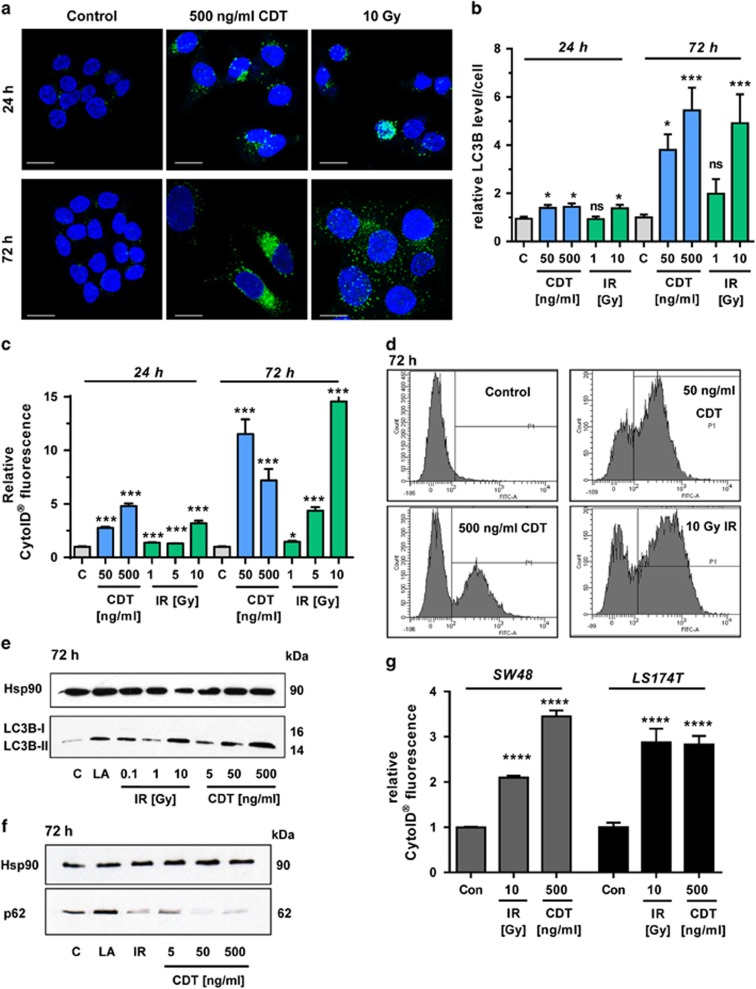
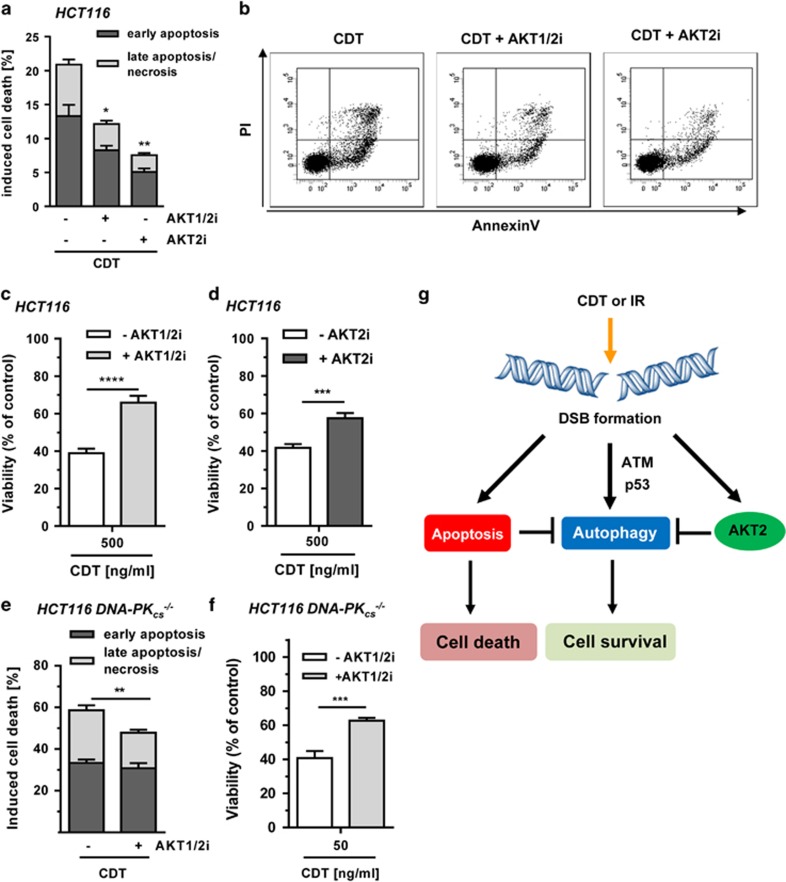
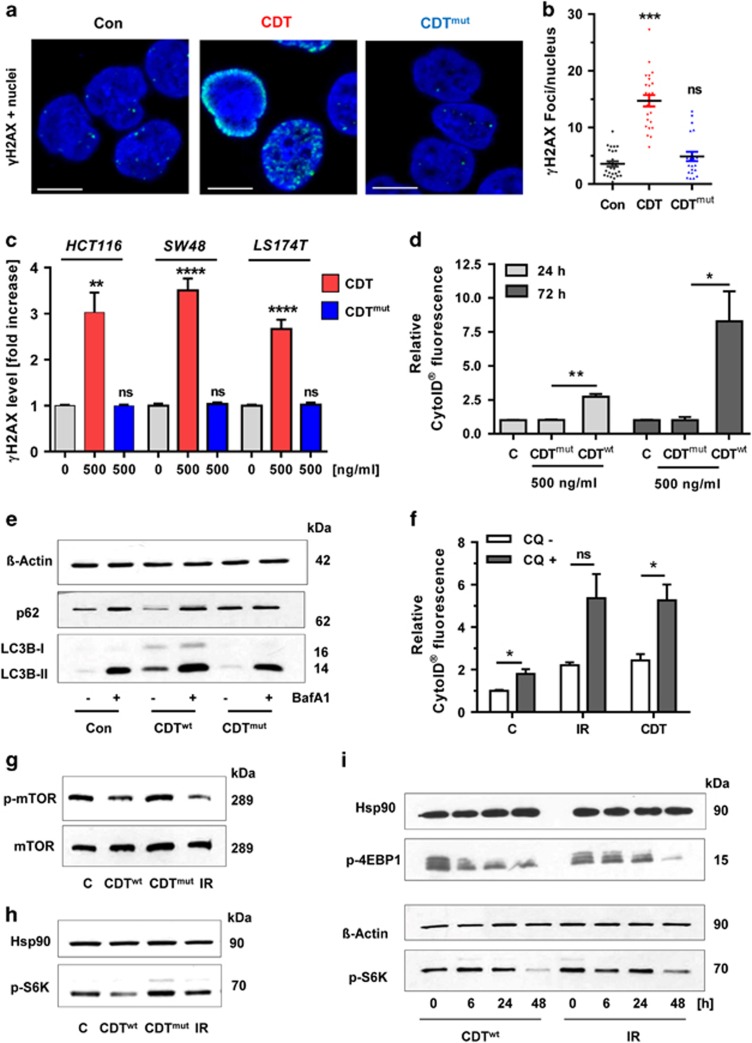
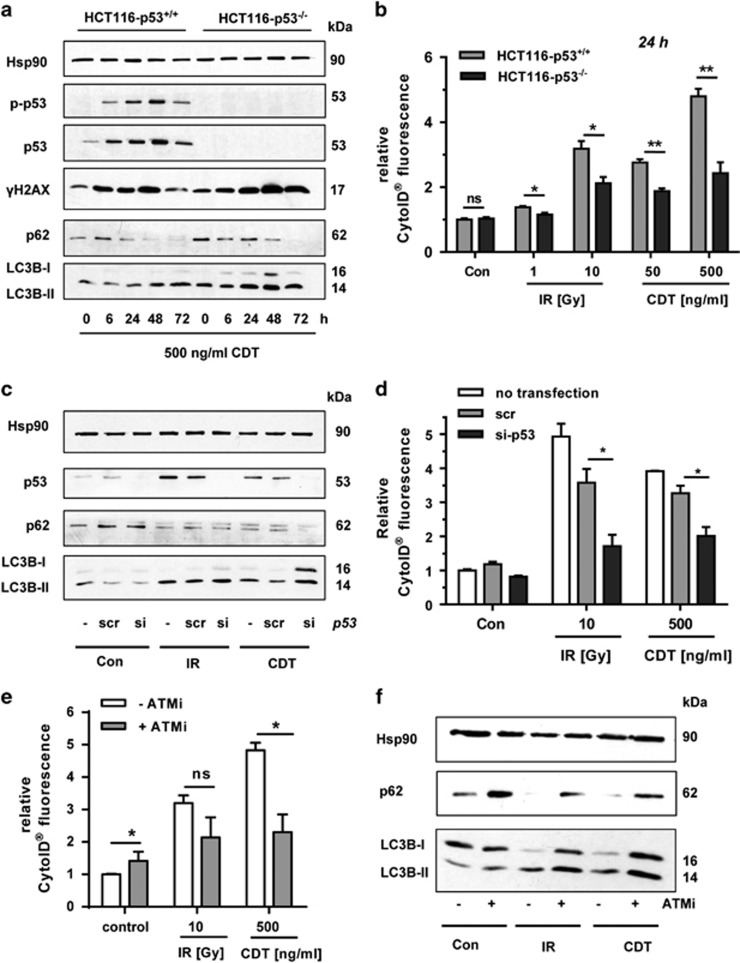
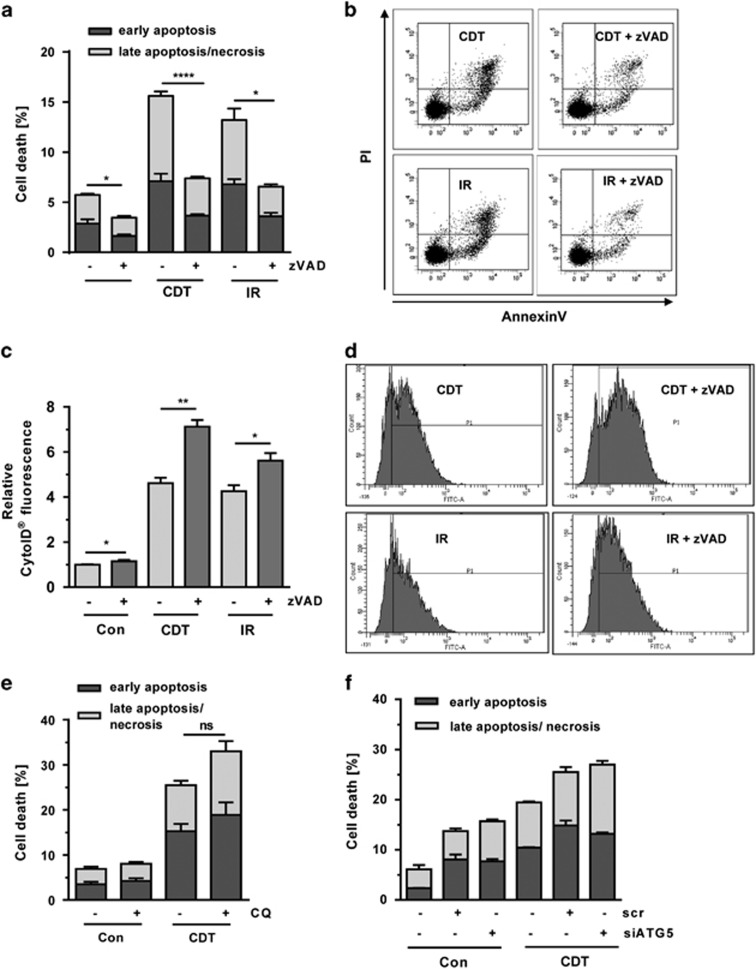
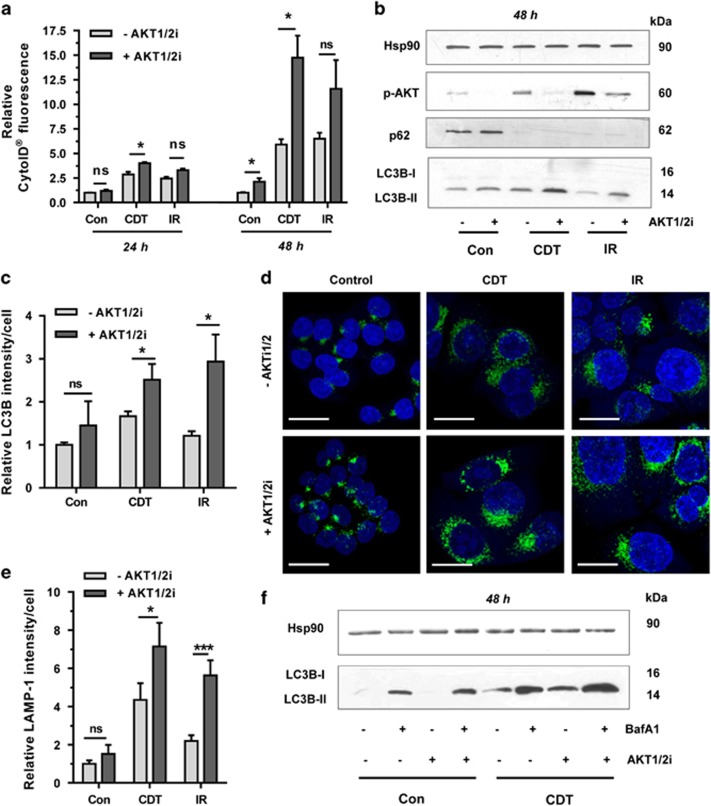
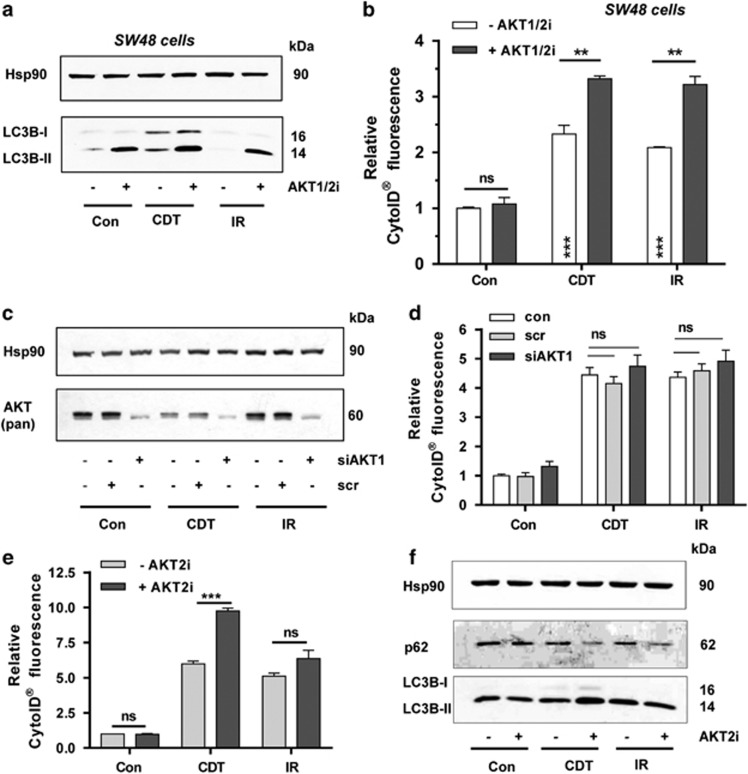
DNA double-strand breaks (DSBs) are critical DNA lesions, which threaten genome stability and cell survival. DSBs are directly induced by ionizing radiation (IR) and radiomimetic agents, including the cytolethal distending toxin (CDT). This bacterial genotoxin harbors a unique DNase-I-like endonuclease activity. Here we studied the role of DSBs induced by CDT and IR as a trigger of autophagy, which is a cellular degradation process involved in cell homeostasis, genome protection and cancer. The regulatory mechanisms of DSB-induced autophagy were analyzed, focusing on the ATM-p53-mediated DNA damage response and AKT signaling in colorectal cancer cells. We show that treatment of cells with CDT or IR increased the levels of the autophagy marker LC3B-II. Consistently, an enhanced formation of autophagosomes and a decrease of the autophagy substrate p62 were observed. Both CDT and IR concomitantly suppressed mTOR signaling and stimulated the autophagic flux. DSBs were demonstrated as the primary trigger of autophagy using a DNase I-defective CDT mutant, which neither induced DSBs nor autophagy. Genetic abrogation of p53 and inhibition of ATM signaling impaired the autophagic flux as revealed by LC3B-II accumulation and reduced formation of autophagic vesicles. Blocking of DSB-induced apoptotic cell death by the pan-caspase inhibitor Z-VAD stimulated autophagy. In line with this, pharmacological inhibition of autophagy increased cell death, while ATG5 knockdown did not affect cell death after DSB induction. Interestingly, both IR and CDT caused AKT activation, which repressed DSB-triggered autophagy independent of the cellular DNA-PK status. Further knockdown and pharmacological inhibitor experiments provided evidence that the negative autophagy regulation was largely attributable to AKT2. Finally, we show that upregulation of CDT-induced autophagy upon AKT inhibition resulted in lower apoptosis and increased cell viability. Collectively, the findings demonstrate that DSBs trigger pro-survival autophagy in an ATM- and p53-dependent manner, which is curtailed by AKT2 signaling.
DNA 双链断裂 (DSBs) 是一种关键的 DNA 损伤,威胁着基因组的稳定性和细胞的存活。DSBs 是由电离辐射 (IR) 和放射模拟物直接诱导的,包括细胞致死扩张毒素 (CDT)。这种细菌遗传毒素具有独特的 DNA 酶 I 样内切酶活性。在这里,我们研究了 CDT 和 IR 诱导的 DSB 作为自噬的触发因素的作用,自噬是一种涉及细胞内稳态、基因组保护和癌症的细胞降解过程。分析了 DSB 诱导的自噬的调节机制,重点是 ATM-p53 介导的 DNA 损伤反应和 AKT 信号在结直肠癌细胞中的作用。我们表明,用 CDT 或 IR 处理细胞会增加自噬标记物 LC3B-II 的水平。一致地,观察到自噬体的形成增强和自噬底物 p62 的减少。CDT 和 IR 均同时抑制 mTOR 信号并刺激自噬通量。使用缺乏 DNA 酶 I 的 CDT 突变体证明 DSB 是自噬的主要触发因素,该突变体既不能诱导 DSB 也不能诱导自噬。p53 的遗传缺失和 ATM 信号的抑制损害了自噬通量,表现为 LC3B-II 的积累和自噬小泡形成减少。用泛半胱天冬酶抑制剂 Z-VAD 阻断 DSB 诱导的凋亡细胞死亡刺激了自噬。与此一致,用药理学抑制自噬增加了细胞死亡,而 ATG5 的敲低不影响 DSB 诱导后的细胞死亡。有趣的是,IR 和 CDT 均导致 AKT 激活,AKT 独立于细胞 DNA-PK 状态抑制 DSB 触发的自噬。进一步的敲低和药理学抑制剂实验提供了证据表明,负性自噬调节主要归因于 AKT2。最后,我们表明,在 AKT 抑制时,CDT 诱导的自噬的上调导致较低的凋亡和增加的细胞活力。总之,这些发现表明,DSBs 以 ATM 和 p53 依赖的方式触发促生存自噬,而 AKT2 信号抑制了自噬。