Integrative Research Center, Science and Education, Field Museum of Natural History, 1400S Lake Shore Drive, Chicago, IL, 60605, USA.
Department of Biology & M. L. Bean Life Science Museum, Brigham Young University, Provo, UT, 84602, USA.
Sci Rep. 2017 Aug 29;7(1):9884. doi: 10.1038/s41598-017-09906-7.
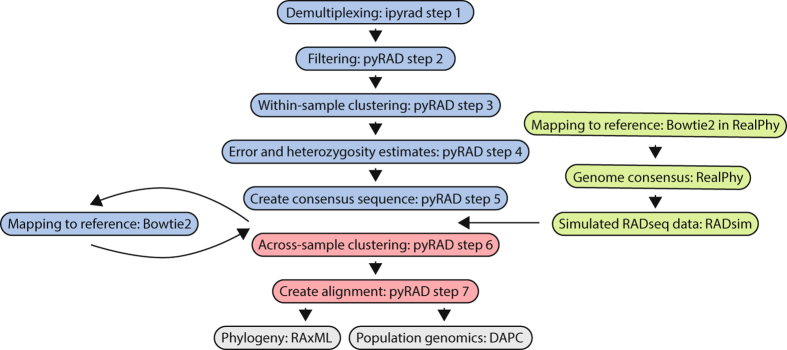
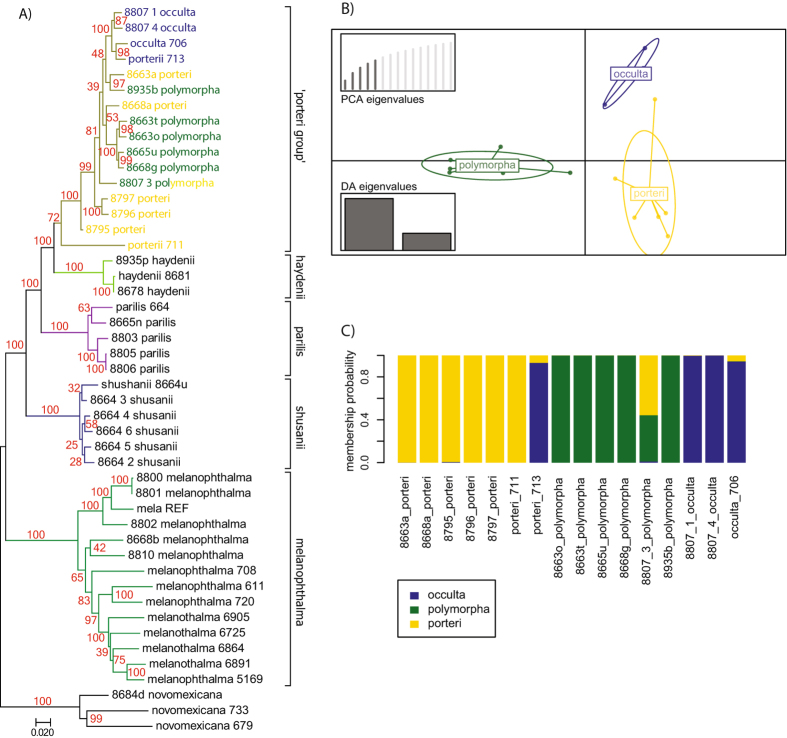
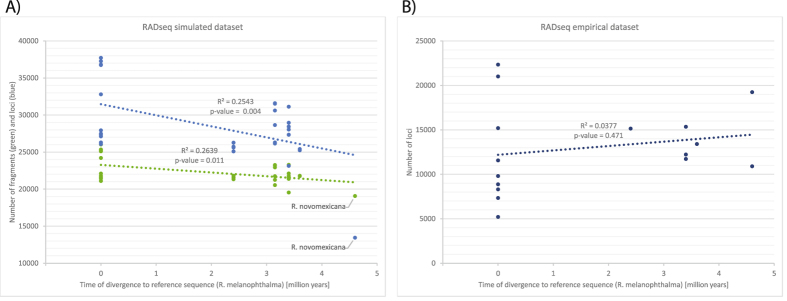
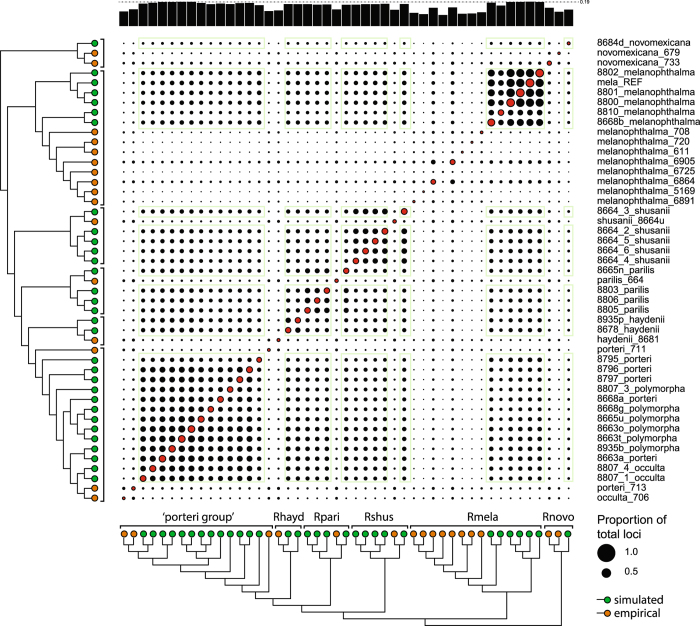
Despite increasing availability of phylogenomic datasets, strategies to generate genome-scale data from organisms involved in symbiotic relationships remains challenging. Restriction site-associated DNA sequencing (RADseq) can effectively generated reduced representation genomic loci. However, when using metagenomic DNA from inseparable symbiotic organisms, RADseq loci may belong to any number of the organisms involved in these intimate associations. In this study, we explored the potential for a reference-based RADseq approach to generate data for lichen-forming fungi from metagenomic DNA extracted from intact lichens. We simulated RAD data from draft genomes of closely related lichenized fungi to test if RADseq can reconstruct robust evolutionary relationships. Subsequently, we generated empirical RADseq data from metagenomic lichen DNA, with RADseq loci mapped back to a reference genome to exclude loci from other lichen symbionts that are represented in metagenomic libraries. In all cases, phylogenetic reconstructions using RADseq loci recovered diversification histories consistent with a previous study based on more comprehensive genome sampling. Furthermore, RADseq loci were found to resolve relationships among closely related species, which were otherwise indistinguishable using a phylogenetic species recognition criterion. Our studies revealed that a modified, reference-based RADseq approach can successfully be implemented to generate symbiont-specific phylogenomic data from metagenomic reads.
尽管越来越多的系统发育基因组数据集可用,但从参与共生关系的生物体中生成基因组规模数据的策略仍然具有挑战性。限制性位点相关 DNA 测序(RADseq)可以有效地生成减少代表性基因组基因座。然而,当使用来自不可分割共生生物体的宏基因组 DNA 时,RADseq 基因座可能属于这些密切相关的共生生物体中的任意数量的生物体。在这项研究中,我们探讨了基于参考的 RADseq 方法从完整地衣中提取的宏基因组 DNA 中生成地衣形成真菌数据的潜力。我们模拟了亲缘关系密切的地衣真菌的草图基因组的 RAD 数据,以测试 RADseq 是否可以重建稳健的进化关系。随后,我们从宏基因组地衣 DNA 中生成了经验性 RADseq 数据,并将 RADseq 基因座映射回参考基因组,以排除宏基因组文库中代表的其他地衣共生体的基因座。在所有情况下,使用 RADseq 基因座进行的系统发育重建都恢复了与先前基于更全面基因组采样的研究一致的多样化历史。此外,RADseq 基因座可用于解决密切相关物种之间的关系,否则使用系统发育种识别标准是无法区分的。我们的研究表明,可以成功实施经过修改的、基于参考的 RADseq 方法,从宏基因组读数中生成共生体特异性的系统基因组数据。