Jorna Jesse, Linde Jackson B, Searle Peter C, Jackson Abigail C, Nielsen Mary-Elise, Nate Madeleine S, Saxton Natalie A, Grewe Felix, Herrera-Campos María de Los Angeles, Spjut Richard W, Wu Huini, Ho Brian, Lumbsch H Thorsten, Leavitt Steven D
Department of Biology Brigham Young University Provo Utah USA.
Science & Education The Grainger Bioinformatics Center The Field Museum Chicago Illinois USA.
Ecol Evol. 2021 Dec 19;11(24):18615-18632. doi: 10.1002/ece3.8467. eCollection 2021 Dec.
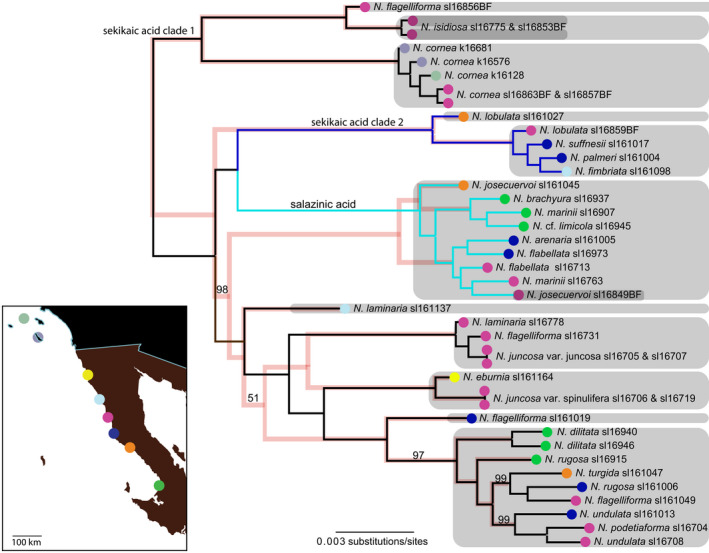
Species delimitation among closely related species is challenging because traditional phenotype-based approaches, for example, using morphology, ecological, or chemical characteristics, may not coincide with natural groupings. With the advent of high-throughput sequencing, it has become increasingly cost-effective to acquire genome-scale data which can resolve previously ambiguous species boundaries. As the availability of genome-scale data has increased, numerous species delimitation analyses, such as BPP and SNAPP+Bayes factor delimitation (BFD*), have been developed to delimit species boundaries. However, even empirical molecular species delimitation approaches can be biased by confounding evolutionary factors, for example, hybridization/introgression and incomplete lineage sorting, and computational limitations. Here, we investigate species boundaries and the potential for micro-endemism in a lineage of lichen-forming fungi, Rundel & Bowler, in the family Ramalinaceae by analyzing single-locus and genome-scale data consisting of (a) single-locus species delimitation analysis using ASAP, (b) maximum likelihood-based phylogenetic tree inference, (c) genome-scale species delimitation models, e.g., BPP and SNAPP+BFD, and (d) species validation using the genealogical divergence index (). We specifically use these methods to cross-validate results between genome-scale and single-locus datasets, differently sampled subsets of genomic data and to control for population-level genetic divergence. Our species delimitation models tend to support more speciose groupings that were inconsistent with traditional taxonomy, supporting a hypothesis of micro-endemism, which may include morphologically cryptic species. However, the models did not converge on robust, consistent species delimitations. While the results of our analysis are somewhat ambiguous in terms of species boundaries, they provide a valuable perspective on how to use these empirical species delimitation methods in a nonmodel system. This study thus highlights the challenges inherent in delimiting species, particularly in groups such as , with complex, relatively recent phylogeographic histories.
在亲缘关系密切的物种之间进行物种界定具有挑战性,因为传统的基于表型的方法,例如使用形态学、生态学或化学特征,可能与自然分组不一致。随着高通量测序的出现,获取能够解决先前模糊物种边界的基因组规模数据变得越来越具有成本效益。随着基因组规模数据可用性的增加,已经开发了许多物种界定分析方法,如贝叶斯泊松树过程(BPP)和贝叶斯因子界定的物种网络分析(SNAPP+BFD*)来界定物种边界。然而,即使是经验性的分子物种界定方法也可能受到混杂的进化因素的影响,例如杂交/基因渗入和不完全谱系分选,以及计算限制。在这里,我们通过分析由以下组成的单基因座和基因组规模数据,研究了鹿角菜科中一个地衣形成真菌谱系(Rundel & Bowler)的物种边界和微特有现象的可能性:(a)使用ASAP进行单基因座物种界定分析,(b)基于最大似然法的系统发育树推断,(c)基因组规模物种界定模型,例如BPP和SNAPP+BFD,以及(d)使用谱系分歧指数()进行物种验证。我们特别使用这些方法来交叉验证基因组规模和单基因座数据集之间的结果、基因组数据的不同采样子集,并控制种群水平的遗传分歧。我们的物种界定模型倾向于支持与传统分类学不一致的更多物种丰富的分组,支持微特有现象的假设,其中可能包括形态上难以区分的物种。然而,这些模型并没有收敛于稳健、一致的物种界定。虽然我们的分析结果在物种边界方面有些模糊,但它们为如何在非模式系统中使用这些经验性物种界定方法提供了有价值的视角。因此,这项研究突出了物种界定中固有的挑战,特别是在具有复杂、相对较新的系统地理学历史的类群中。