Ofaim Shany, Ofek-Lalzar Maya, Sela Noa, Jinag Jiandong, Kashi Yechezkel, Minz Dror, Freilich Shiri
Newe Ya'ar Research Center, Agricultural Research OrganizationRamat Yishay, Israel.
Faculty of Biotechnology and Food Engineering, Technion-Israel Institute of TechnologyHaifa, Israel.
Front Microbiol. 2017 Aug 23;8:1606. doi: 10.3389/fmicb.2017.01606. eCollection 2017.
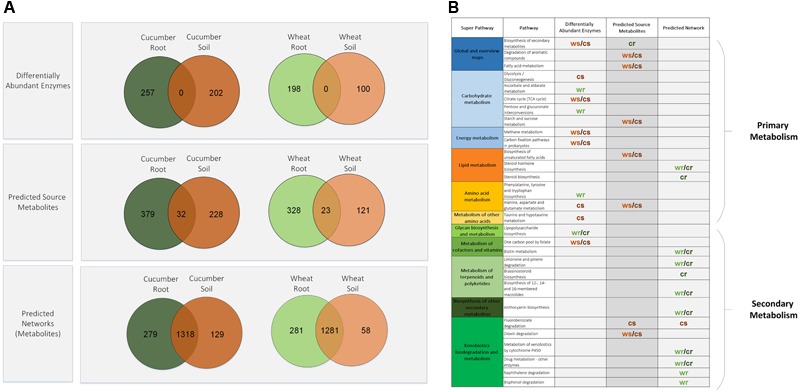
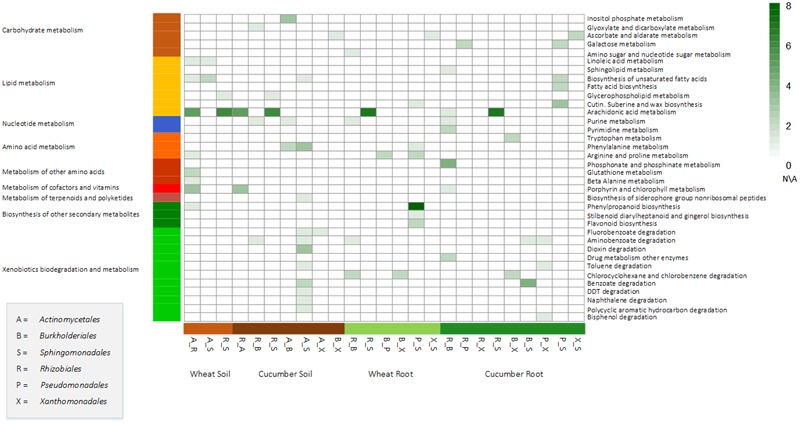
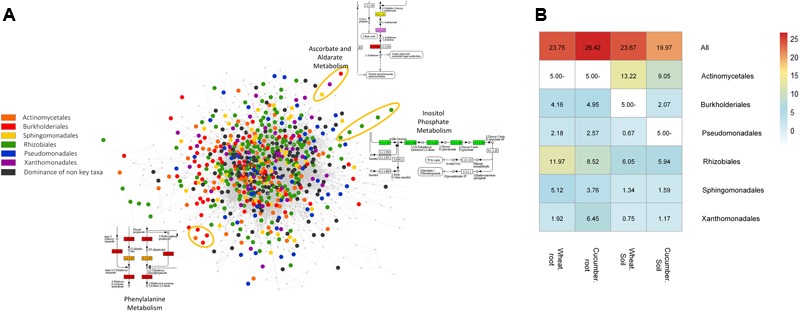
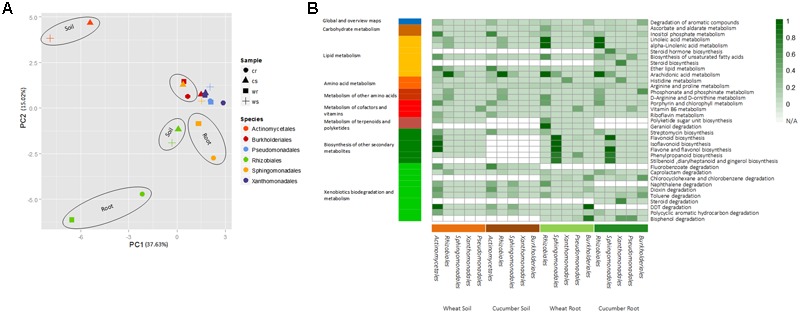
Advances in metagenomics enable high resolution description of complex bacterial communities in their natural environments. Consequently, conceptual approaches for community level functional analysis are in high need. Here, we introduce a framework for a metagenomics-based analysis of community functions. Environment-specific gene catalogs, derived from metagenomes, are processed into metabolic-network representation. By applying established ecological conventions, network-edges (metabolic functions) are assigned with taxonomic annotations according to the dominance level of specific groups. Once a function-taxonomy link is established, prediction of the impact of dominant taxa on the overall community performances is assessed by simulating removal or addition of edges (taxa associated functions). This approach is demonstrated on metagenomic data describing the microbial communities from the root environment of two crop plants - wheat and cucumber. Predictions for environment-dependent effects revealed differences between treatments (root vs. soil), corresponding to documented observations. Metabolism of specific plant exudates (e.g., organic acids, flavonoids) was linked with distinct taxonomic groups in simulated root, but not soil, environments. These dependencies point to the impact of these metabolite families as determinants of community structure. Simulations of the activity of pairwise combinations of taxonomic groups (order level) predicted the possible production of complementary metabolites. Complementation profiles allow formulating a possible metabolic role for observed co-occurrence patterns. For example, production of tryptophan-associated metabolites through complementary interactions is unique to the tryptophan-deficient cucumber root environment. Our approach enables formulation of testable predictions for species contribution to community activity and exploration of the functional outcome of structural shifts in complex bacterial communities. Understanding community-level metabolism is an essential step toward the manipulation and optimization of microbial function. Here, we introduce an analysis framework addressing three key challenges of such data: producing quantified links between taxonomy and function; contextualizing discrete functions into communal networks; and simulating environmental impact on community performances. New technologies will soon provide a high-coverage description of biotic and a-biotic aspects of complex microbial communities such as these found in gut and soil. This framework was designed to allow the integration of high-throughput metabolomic and metagenomic data toward tackling the intricate associations between community structure, community function, and metabolic inputs.
宏基因组学的进展使得在自然环境中对复杂细菌群落进行高分辨率描述成为可能。因此,对群落水平功能分析的概念性方法有着迫切需求。在此,我们介绍一种基于宏基因组学的群落功能分析框架。从宏基因组中获得的特定环境基因目录被处理成代谢网络表示形式。通过应用既定的生态学惯例,根据特定群体的优势水平为网络边(代谢功能)赋予分类注释。一旦建立了功能 - 分类学联系,就通过模拟去除或添加边(与分类群相关的功能)来评估优势分类群对整体群落性能的影响。这种方法在描述两种农作物(小麦和黄瓜)根际环境中微生物群落的宏基因组数据上得到了验证。对环境依赖性效应的预测揭示了处理方式(根际与土壤)之间的差异,这与已记录的观察结果相符。在模拟的根际环境而非土壤环境中,特定植物分泌物(如有机酸、黄酮类化合物)的代谢与不同的分类群相关联。这些依赖性表明这些代谢物家族作为群落结构决定因素的影响。对分类群(目水平)成对组合活性的模拟预测了互补代谢物的可能产生。互补图谱有助于为观察到的共现模式制定可能的代谢作用。例如,通过互补相互作用产生色氨酸相关代谢物是色氨酸缺乏的黄瓜根际环境所特有的。我们的方法能够对物种对群落活动的贡献做出可测试的预测,并探索复杂细菌群落结构变化的功能结果。理解群落水平的代谢是操纵和优化微生物功能的关键一步。在此,我们介绍一个分析框架,该框架解决了此类数据的三个关键挑战:在分类学和功能之间建立量化联系;将离散功能置于群落网络中进行情境化;以及模拟环境对群落性能的影响。新技术很快将对复杂微生物群落(如在肠道和土壤中发现的那些)的生物和非生物方面进行高覆盖率描述。这个框架旨在允许整合高通量代谢组学和宏基因组学数据,以解决群落结构、群落功能和代谢输入之间的复杂关联。