Hedaya Mohsen A, Thomas Vidhya, Abdel-Hamid Mohamed E, Kehinde Elijah O, Phillips Oludotun A
Department of Pharmaceutics, Faculty of Pharmacy, Kuwait University, P.O. Box 24923, Safat 13110, Kuwait.
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Kuwait University, P.O. Box 24923, Safat 13110, Kuwait.
Pharmaceutics. 2017 Sep 7;9(3):34. doi: 10.3390/pharmaceutics9030034.
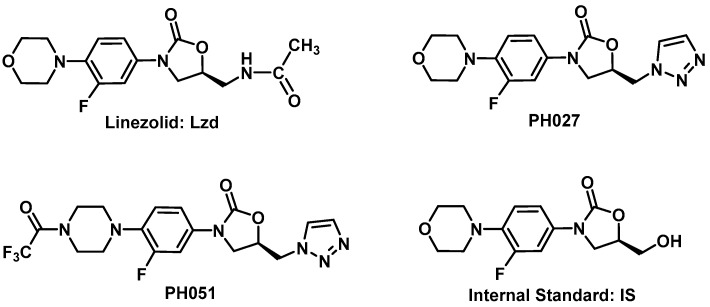
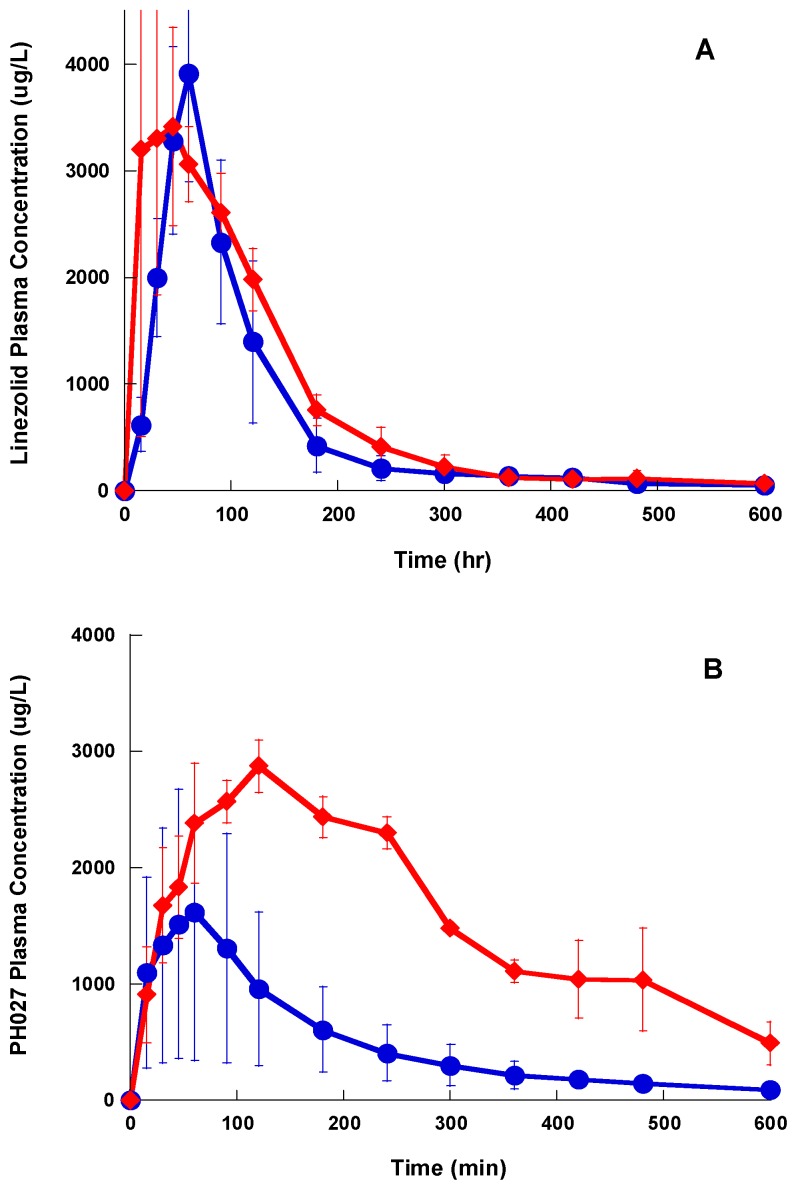
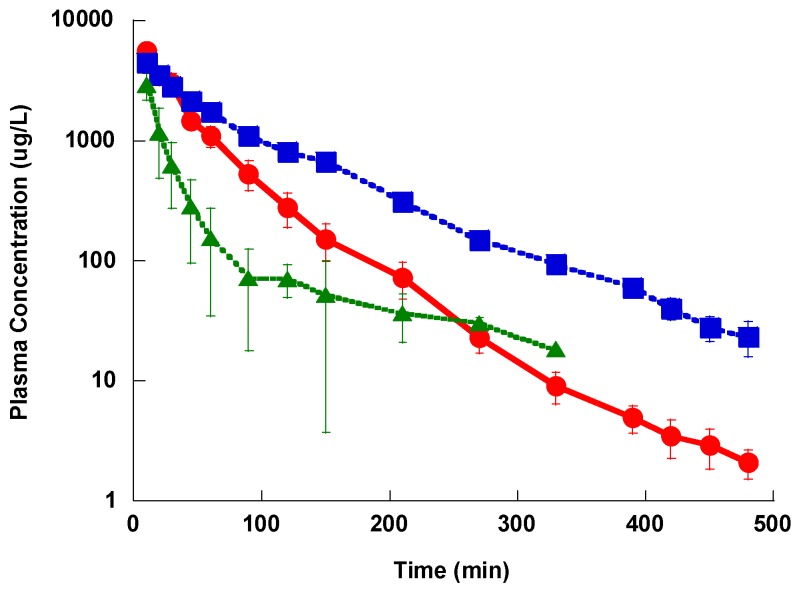
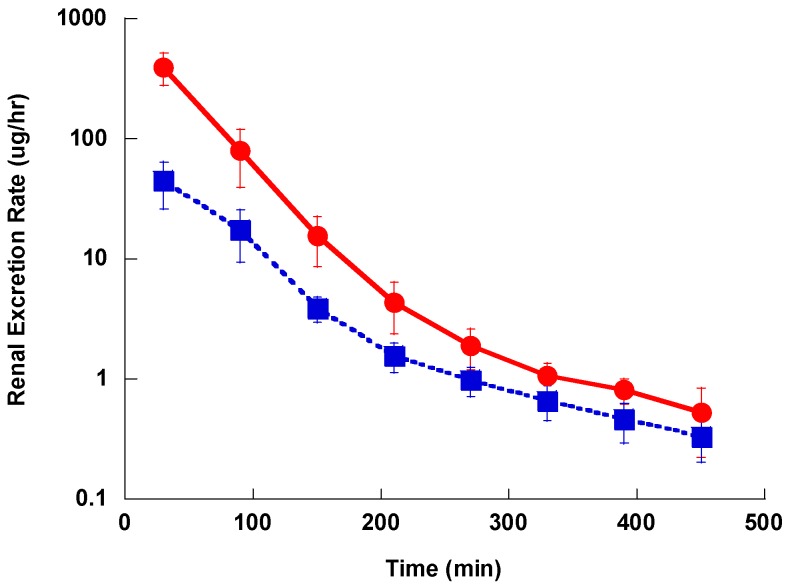
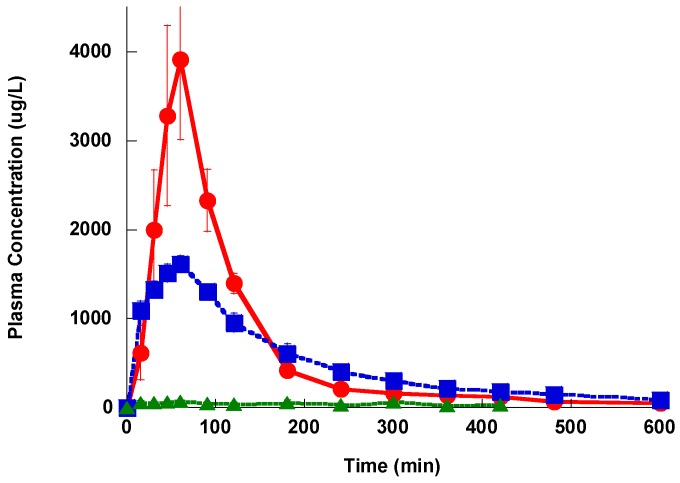
This is a comparative pharmacokinetics study of linezolid (Lzd), and two novel oxazolidinone antibacterial agents-PH027 and PH051-in rabbits to determine if the discrepancy between the in vitro and in vivo activities of the novel compounds is due to pharmacokinetic factors. The pharmacokinetics after IV and oral administration, plasma protein binding and tissue distribution for the three compounds were compared. The elimination half-lives were 52.4 ± 6.3, 68.7 ± 12.1 and 175 ± 46.1 min for Lzd, PH027 and PH051, respectively. The oral bioavailability for Lzd, PH027 and PH051 administered as suspension were 38.7%, 22.1% and 4.73%, which increased significantly when administered as microemulsion to 51.7%, 72.9% and 13.9%. The plasma protein binding were 32-34%, 37-38% and 90-91% for Lzd, PH027 and PH051. The tissue distribution for PH027 and PH051 in all investigated tissues were higher than that for Lzd. It can be concluded that the lower bioavailability of PH027 and PH051 compared to Lzd when administered as suspension is the main cause of their lower in vivo activity, despite their comparable in vitro activity. Differences in the other pharmacokinetic characteristics cannot explain the lower in vivo activity. The in vivo activity of the novel compounds should be re-evaluated using formulations with good oral bioavailability.
这是一项关于利奈唑胺(Lzd)以及两种新型恶唑烷酮类抗菌剂——PH027和PH051在兔体内的比较药代动力学研究,以确定新型化合物体外和体内活性之间的差异是否归因于药代动力学因素。比较了三种化合物静脉注射和口服给药后的药代动力学、血浆蛋白结合率和组织分布情况。Lzd、PH027和PH051的消除半衰期分别为52.4±6.3、68.7±12.1和175±46.1分钟。以混悬液形式给药时,Lzd、PH027和PH051的口服生物利用度分别为38.7%、22.1%和4.73%,当以微乳剂形式给药时,生物利用度显著提高至51.7%、72.9%和13.9%。Lzd、PH027和PH051的血浆蛋白结合率分别为32 - 34%、37 - 38%和90 - 91%。PH027和PH051在所有研究组织中的组织分布均高于Lzd。可以得出结论,尽管PH027和PH051与Lzd体外活性相当,但以混悬液形式给药时,它们的生物利用度低于Lzd是其体内活性较低的主要原因。其他药代动力学特征的差异无法解释其较低的体内活性。应使用具有良好口服生物利用度的制剂重新评估新型化合物的体内活性。