Department of Computer Science, Stony Brook University, Stony Brook, NY, USA.
VMWare, Inc., Palo Alto, CA.
Bioinformatics. 2017 Jul 15;33(14):i133-i141. doi: 10.1093/bioinformatics/btx261.
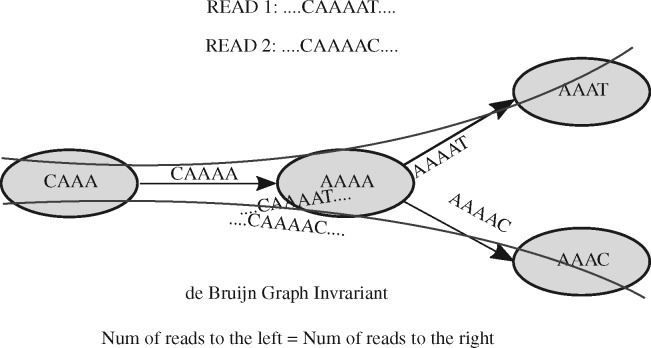
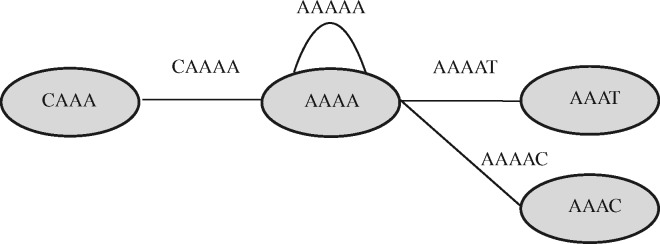
Almost all de novo short-read genome and transcriptome assemblers start by building a representation of the de Bruijn Graph of the reads they are given as input. Even when other approaches are used for subsequent assembly (e.g. when one is using 'long read' technologies like those offered by PacBio or Oxford Nanopore), efficient k -mer processing is still crucial for accurate assembly, and state-of-the-art long-read error-correction methods use de Bruijn Graphs. Because of the centrality of de Bruijn Graphs, researchers have proposed numerous methods for representing de Bruijn Graphs compactly. Some of these proposals sacrifice accuracy to save space. Further, none of these methods store abundance information, i.e. the number of times that each k -mer occurs, which is key in transcriptome assemblers.
We present a method for compactly representing the weighted de Bruijn Graph (i.e. with abundance information) with essentially no errors. Our representation yields zero errors while increasing the space requirements by less than 18-28% compared to the approximate de Bruijn graph representation in Squeakr. Our technique is based on a simple invariant that all weighted de Bruijn Graphs must satisfy, and hence is likely to be of general interest and applicable in most weighted de Bruijn Graph-based systems.
https://github.com/splatlab/debgr .
Supplementary data are available at Bioinformatics online.
几乎所有从头开始的短读基因组和转录组装配器都从构建输入的读取的 de Bruijn 图的表示开始。即使在后续组装中使用其他方法(例如,当使用 PacBio 或 Oxford Nanopore 等“长读”技术时),高效的 k-mer 处理对于准确组装仍然至关重要,并且最先进的长读纠错方法使用 de Bruijn 图。由于 de Bruijn 图的中心性,研究人员已经提出了许多方法来紧凑地表示 de Bruijn 图。其中一些提案为了节省空间而牺牲了准确性。此外,这些方法都没有存储丰度信息,即每个 k-mer 出现的次数,这在转录组装配器中是关键。
我们提出了一种紧凑表示加权 de Bruijn 图(即具有丰度信息)的方法,几乎没有错误。与 Squeakr 中的近似 de Bruijn 图表示相比,我们的表示方法增加的空间需求不到 18-28%,同时增加的错误为零。我们的技术基于所有加权 de Bruijn 图都必须满足的简单不变量,因此可能具有普遍的兴趣并且适用于大多数基于加权 de Bruijn 图的系统。
https://github.com/splatlab/debgr。
补充数据可在 Bioinformatics 在线获取。