Dufault-Thompson Keith, Jiang Xiaofang
Intramural Research Program National Library of Medicine, National Institutes of Health Bethesda Maryland USA.
Imeta. 2022 Mar 1;1(1):e4. doi: 10.1002/imt2.4. eCollection 2022 Mar.
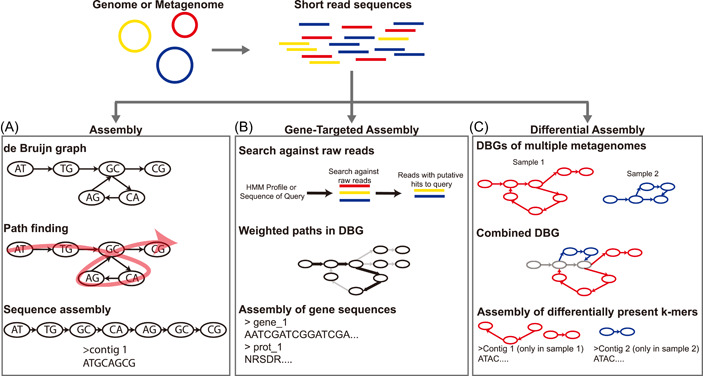
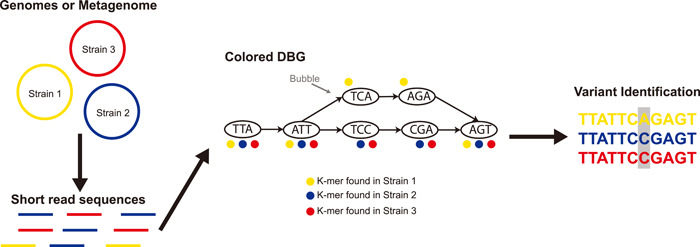
High-throughput sequencing has become an increasingly central component of microbiome research. The development of de Bruijn graph-based methods for assembling high-throughput sequencing data has been an important part of the broader adoption of sequencing as part of biological studies. Recent advances in the construction and representation of de Bruijn graphs have led to new approaches that utilize the de Bruijn graph data structure to aid in different biological analyses. One type of application of these methods has been in alternative approaches to the assembly of sequencing data like gene-targeted assembly, where only gene sequences are assembled out of larger metagenomes, and differential assembly, where sequences that are differentially present between two samples are assembled. de Bruijn graphs have also been applied for comparative genomics where they can be used to represent large sets of multiple genomes or metagenomes where structural features in the graphs can be used to identify variants, indels, and homologous regions in sequences. These de Bruijn graph-based representations of sequencing data have even begun to be applied to whole sequencing databases for large-scale searches and experiment discovery. de Bruijn graphs have played a central role in how high-throughput sequencing data is worked with, and the rapid development of new tools that rely on these data structures suggests that they will continue to play an important role in biology in the future.
高通量测序已日益成为微生物组研究的核心组成部分。基于德布鲁因图的高通量测序数据组装方法的发展,是测序作为生物学研究一部分得以更广泛应用的重要组成部分。德布鲁因图构建和表示方面的最新进展催生了新方法,这些方法利用德布鲁因图数据结构辅助进行不同的生物学分析。这些方法的一类应用是在测序数据组装的替代方法中,如基因靶向组装(仅从较大的宏基因组中组装基因序列)和差异组装(组装两个样本间差异存在的序列)。德布鲁因图也已应用于比较基因组学,在其中可用于表示大量多个基因组或宏基因组,图中的结构特征可用于识别序列中的变异、插入缺失和同源区域。这些基于德布鲁因图的测序数据表示甚至已开始应用于整个测序数据库,用于大规模搜索和实验发现。德布鲁因图在处理高通量测序数据的方式中发挥了核心作用,而依赖这些数据结构的新工具的快速发展表明,它们未来将继续在生物学中发挥重要作用。