Department of Genetics, Rutgers University, Piscataway, NJ, USA.
Human Genetics Institute of New Jersey, Piscataway, NJ, USA.
Mol Psychiatry. 2018 Jun;23(6):1487-1495. doi: 10.1038/mp.2017.179. Epub 2017 Sep 12.
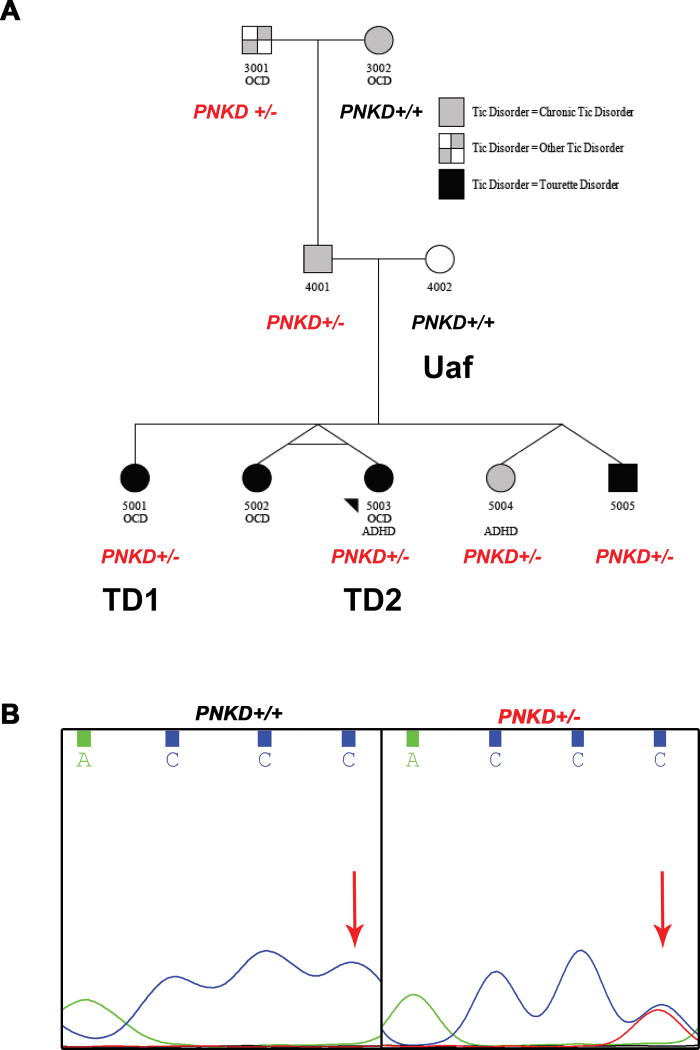
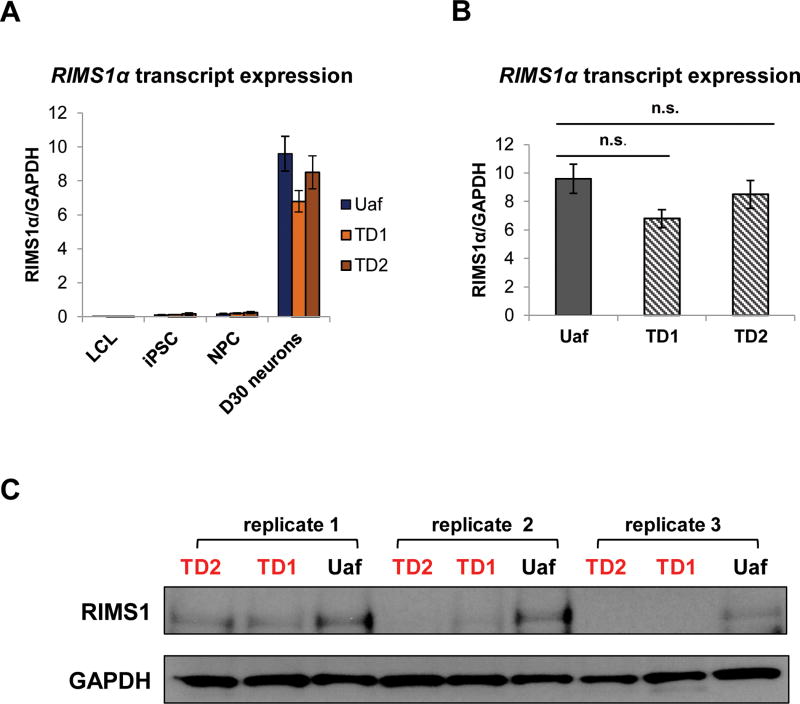
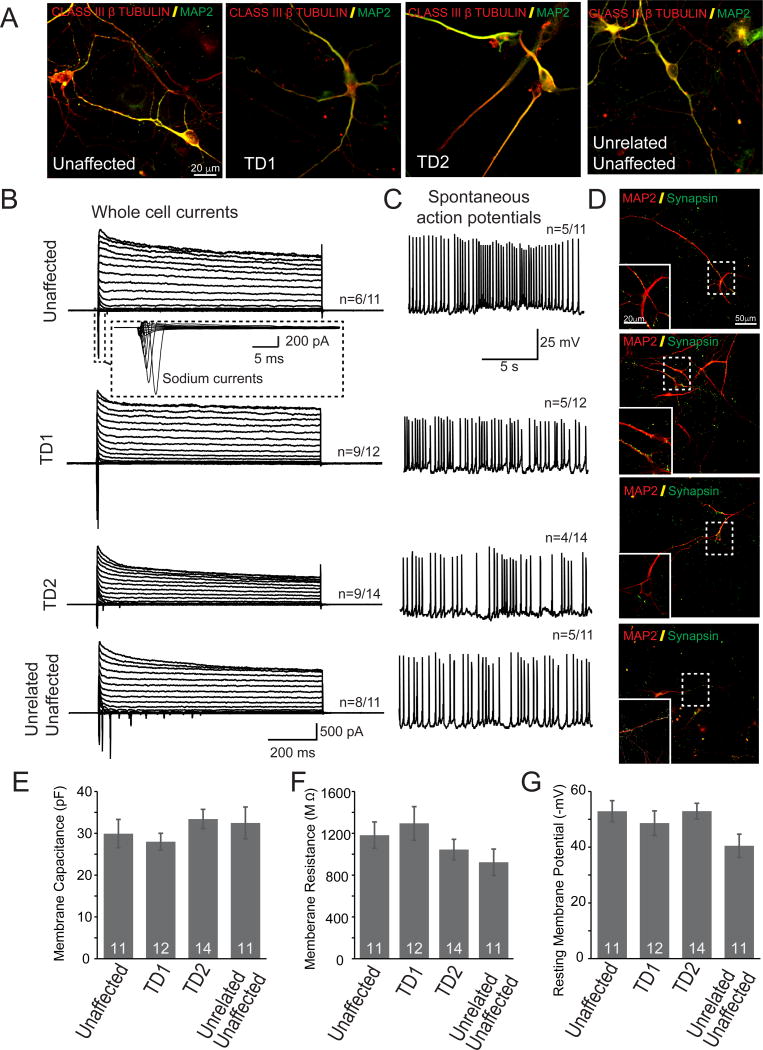
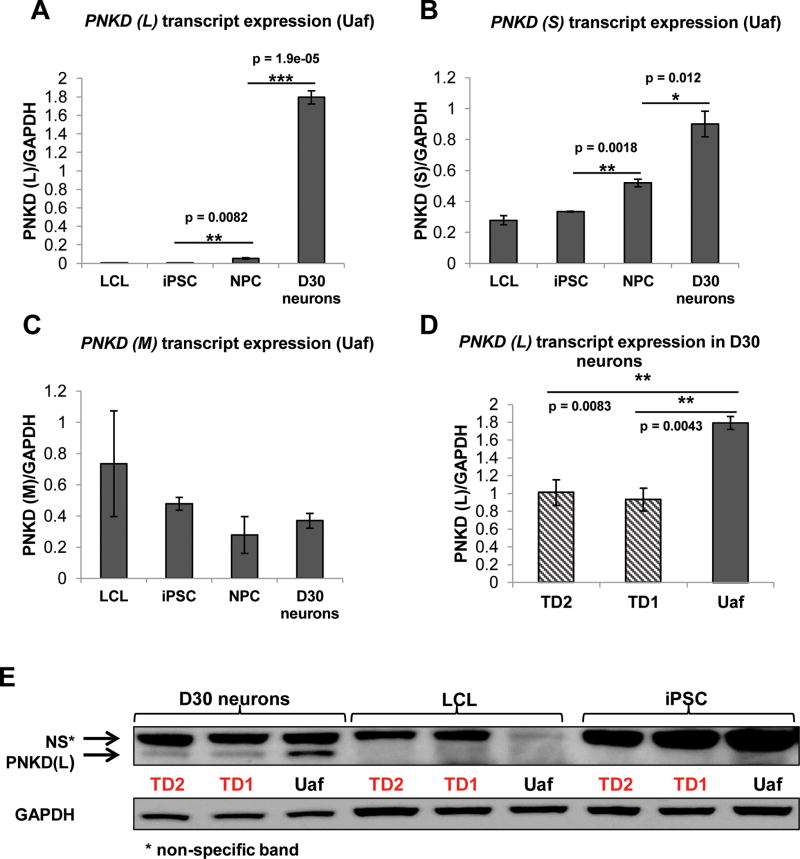
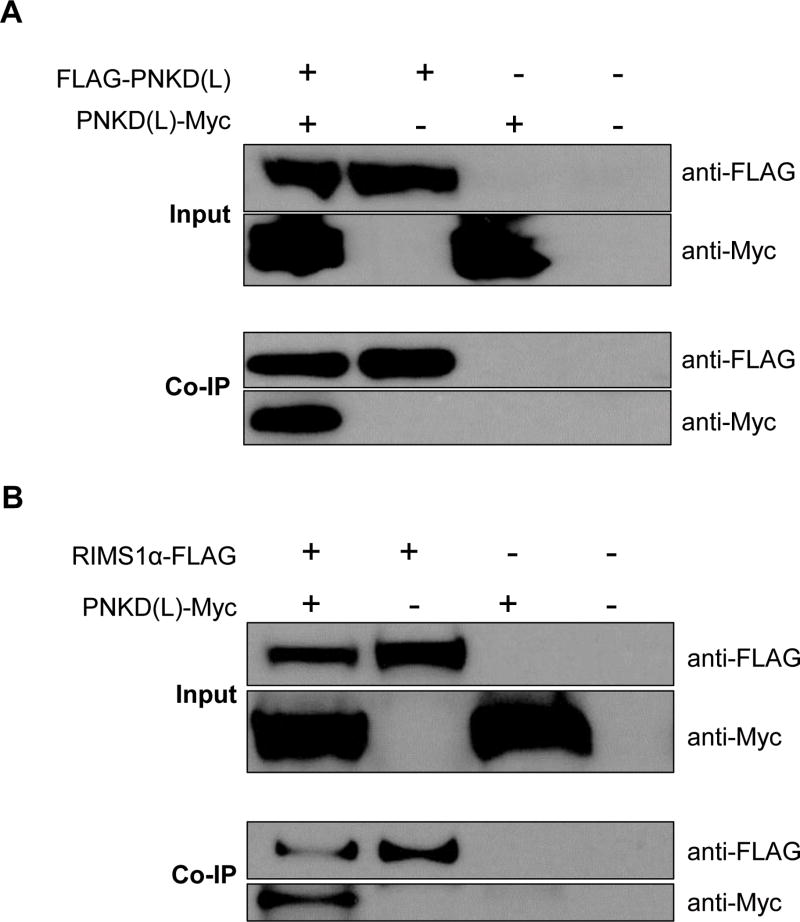
Tourette Disorder (TD) is a childhood-onset neuropsychiatric and neurodevelopmental disorder characterized by the presence of both motor and vocal tics. The genetic architecture of TD is believed to be complex and heterogeneous. Nevertheless, DNA sequence variants co-segregating with TD phenotypes within multiplex families have been identified. This report examines whole exomes of affected and unaffected individuals in a multiplex TD family to discover genes involved in the TD etiology. We performed whole exome sequencing on six out of nine members in a three-generation TD multiplex family. Putative deleterious sequence variants co-segregating with TD patients were identified by our in-house bioinformatics pipeline. Induced pluripotent stem cells (iPSCs) were generated from one unaffected and two TD affected individuals. Neurons were derived from the iPSCs and biochemical assays were conducted to evaluate possible molecular differences between affected and unaffected. A rare heterozygous nonsense mutation in PNKD was co-segregated with TD in this multiplex family. Transcript and protein levels of the PNKD long isoform were reduced in neurons derived from the individuals with TD due to the nonsense mutation, indicating nonsense-mediated mRNA decay. We demonstrated that the PNKD long isoform monomer oligomerizes with itself as well as interacts with the synaptic active zone protein RIMS1α. We concluded that reduced PNKD long isoform levels are detected in all affected individuals and we provide evidence for a mechanism whereby this might contribute to the TD phenotype.
妥瑞氏症(TD)是一种儿童期发病的神经精神和神经发育障碍,其特征是存在运动性和发声性抽动。TD 的遗传结构被认为是复杂和异质的。然而,在多基因家族中,与 TD 表型共分离的 DNA 序列变异已经被确定。本报告研究了一个多基因 TD 家族中受影响和未受影响个体的外显子组,以发现与 TD 病因相关的基因。我们对一个三代 TD 多基因家族中的九名成员中的六名进行了外显子组测序。通过我们内部的生物信息学管道,鉴定出与 TD 患者共分离的潜在有害序列变异。从一个未受影响和两个 TD 受影响的个体中生成诱导多能干细胞(iPSC)。从 iPSC 中衍生出神经元,并进行生化分析,以评估受影响和未受影响个体之间可能存在的分子差异。在这个多基因家族中,PNKD 中的一个罕见杂合无义突变与 TD 共分离。由于无义突变,PNKD 长异构体的转录和蛋白水平在源自 TD 患者的神经元中降低,表明无义介导的 mRNA 衰减。我们证明了 PNKD 长异构体单体自身聚合,以及与突触活性区蛋白 RIMS1α相互作用。我们得出结论,所有受影响的个体中都检测到 PNKD 长异构体水平降低,并且我们提供了证据表明这可能导致 TD 表型的机制。