Department of Orthopaedics, Changzheng Hospital Αffiliated to Second Military Medical University, Shanghai 200003, P.R. China.
Int J Mol Med. 2017 Nov;40(5):1357-1364. doi: 10.3892/ijmm.2017.3126. Epub 2017 Sep 7.
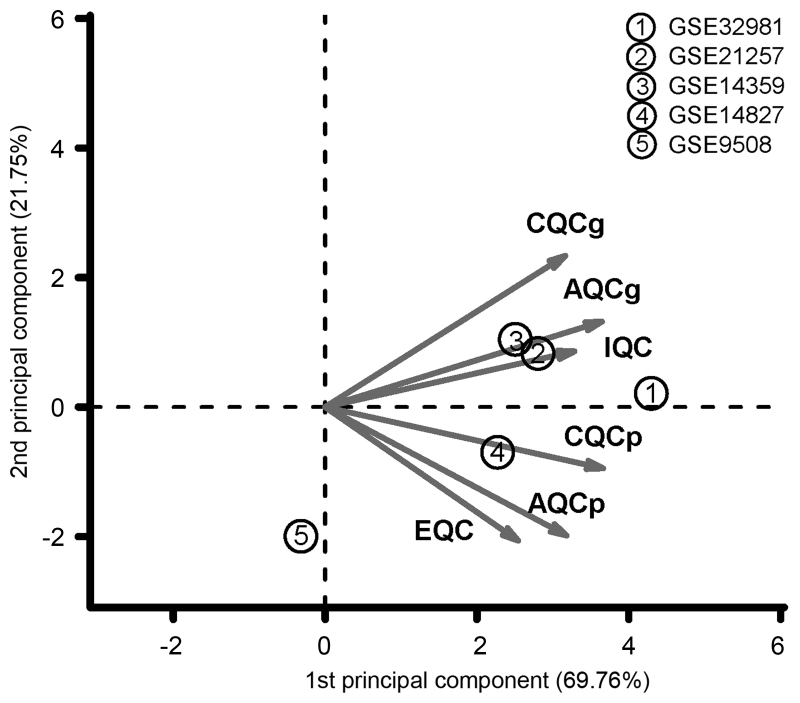
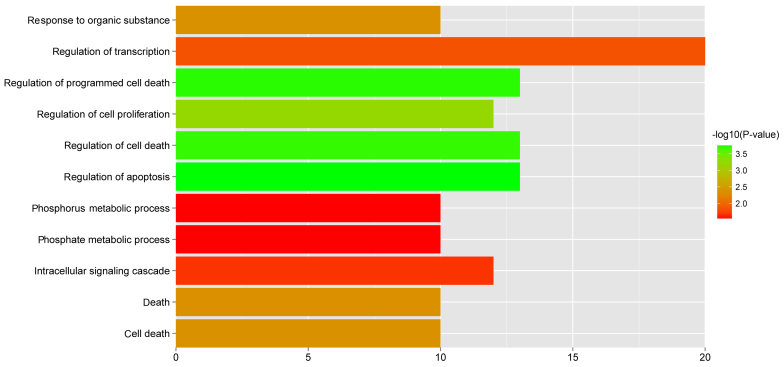
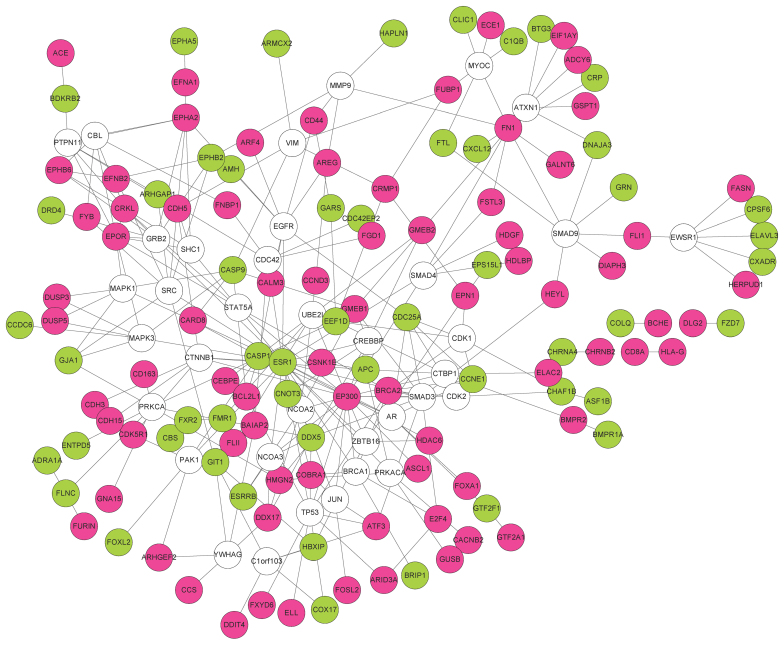
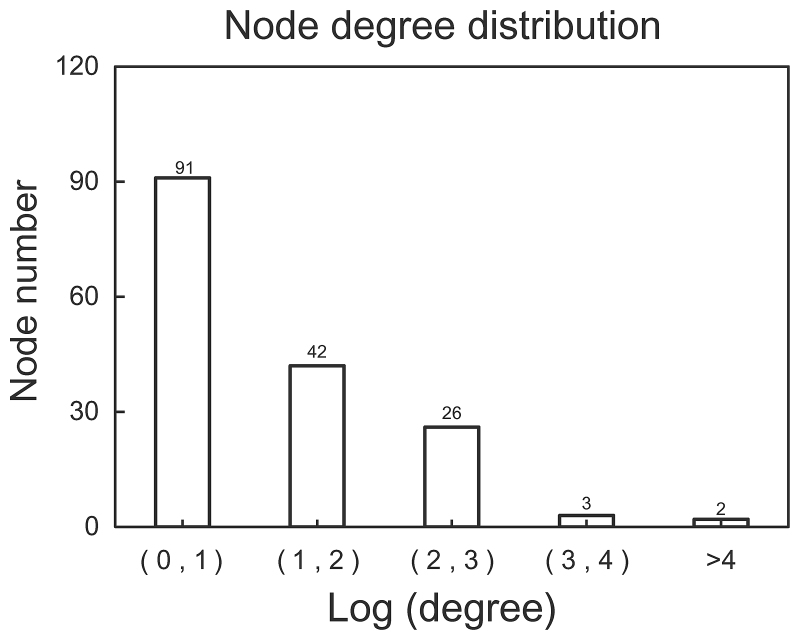
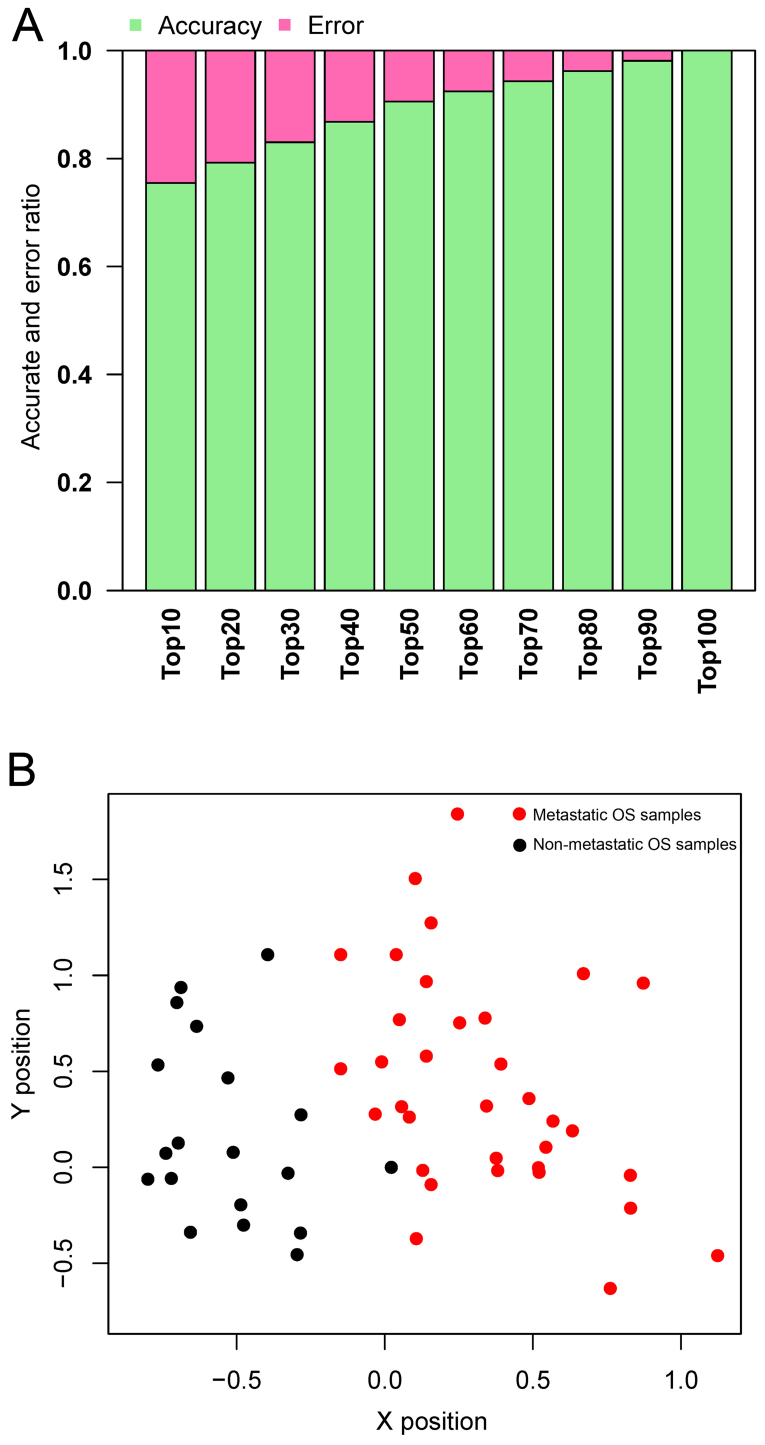
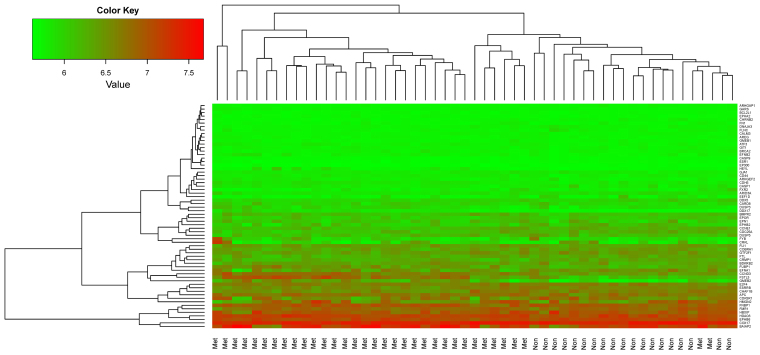
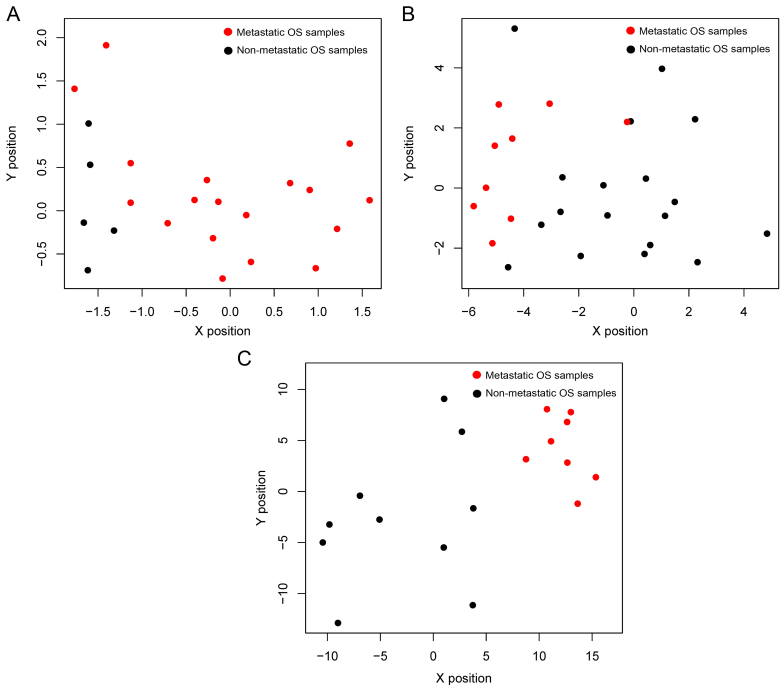
In this study, gene expression profiles of osteosarcoma (OS) were analyzed to identify critical genes associated with metastasis. Five gene expression datasets were screened and downloaded from Gene Expression Omnibus (GEO). Following assessment by MetaQC, the dataset GSE9508 was excluded for poor quality. Subsequently, differentially expressed genes (DEGs) between metastatic and non-metastatic OS were identified using meta‑analysis. A protein-protein interaction (PPI) network was constructed with information from Human Protein Reference Database (HPRD) for the DEGs. Betweenness centrality (BC) was calculated for each node in the network and top featured genes ranked by BC were selected out to construct support vector machine (SVM) classifier using the training set GSE21257, which was then validated using the other three independent datasets. Pathway enrichment analysis was performed for the featured genes using Fisher's exact test. A total of 353 DEGs were identified and a PPI network including 164 nodes and 272 edges was then constructed. The top 64 featured genes ranked by BC were included in the SVM classifier. The SVM classifier exhibited high prediction accuracies in all of the 4 datasets, with accuracies of 100, 100, 92.6 and 100%, respectively. Further analysis of the featured genes revealed that 11 Gene Ontology (GO) biological pathways and 5 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were significantly over-represented, including the regulation of cell proliferation, regulation of apoptosis, pathways in cancer, regulation of actin cytoskeleton and the TGF-β signaling pathway. On the whole, an SVM classifier with high prediction accuracy was constructed and validated, in which key genes associated with metastasis in OS were also revealed. These findings may promote the development of genetic diagnostic methods and may enhance our understanding of the molecular mechanisms underlying the metastasis of OS.
在这项研究中,分析了骨肉瘤(OS)的基因表达谱,以鉴定与转移相关的关键基因。从基因表达综合数据库(GEO)筛选并下载了五个基因表达数据集。经过 MetaQC 评估,数据集 GSE9508 因质量差而被排除。随后,使用荟萃分析鉴定转移性和非转移性 OS 之间的差异表达基因(DEGs)。使用 Human Protein Reference Database(HPRD)中的信息构建了 DEGs 的蛋白质-蛋白质相互作用(PPI)网络。计算网络中每个节点的介数中心度(BC),并选择 BC 排名靠前的特征基因用于构建支持向量机(SVM)分类器,使用训练集 GSE21257,然后使用另外三个独立数据集进行验证。Fisher 精确检验用于对特征基因进行通路富集分析。共鉴定出 353 个 DEG,构建了一个包含 164 个节点和 272 个边的 PPI 网络。BC 排名前 64 的特征基因被纳入 SVM 分类器。SVM 分类器在所有 4 个数据集的预测准确率均较高,分别为 100%、100%、92.6%和 100%。对特征基因的进一步分析表明,11 个基因本体论(GO)生物学途径和 5 个京都基因与基因组百科全书(KEGG)途径显著过度表达,包括细胞增殖的调节、细胞凋亡的调节、癌症途径、肌动蛋白细胞骨架的调节和 TGF-β 信号通路。总的来说,构建并验证了一个预测准确率较高的 SVM 分类器,其中还揭示了与 OS 转移相关的关键基因。这些发现可能促进遗传诊断方法的发展,并增强我们对 OS 转移分子机制的理解。