Mägi Reedik, Horikoshi Momoko, Sofer Tamar, Mahajan Anubha, Kitajima Hidetoshi, Franceschini Nora, McCarthy Mark I, Morris Andrew P
Estonian Genome Center, University of Tartu, Tartu, Estonia.
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK.
Hum Mol Genet. 2017 Sep 15;26(18):3639-3650. doi: 10.1093/hmg/ddx280.
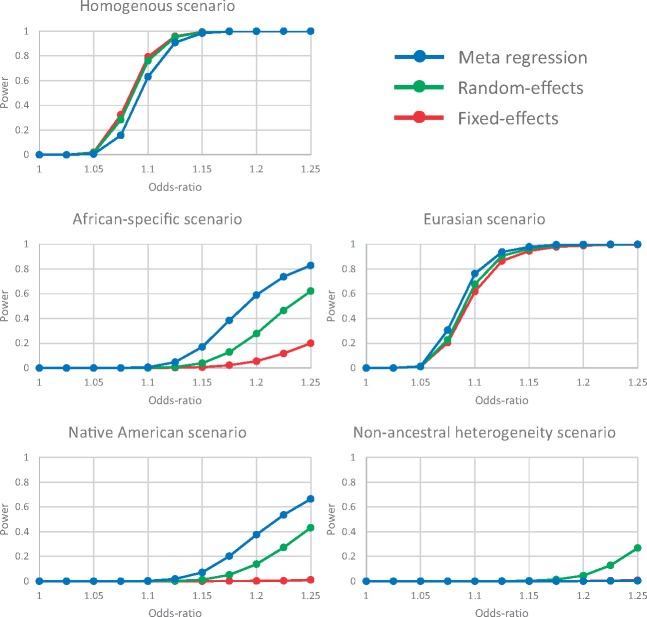
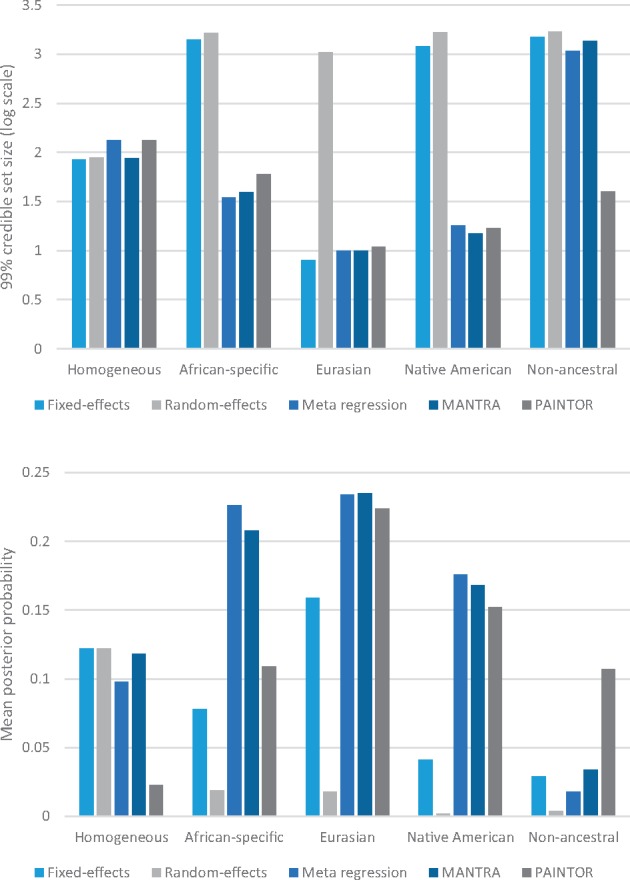
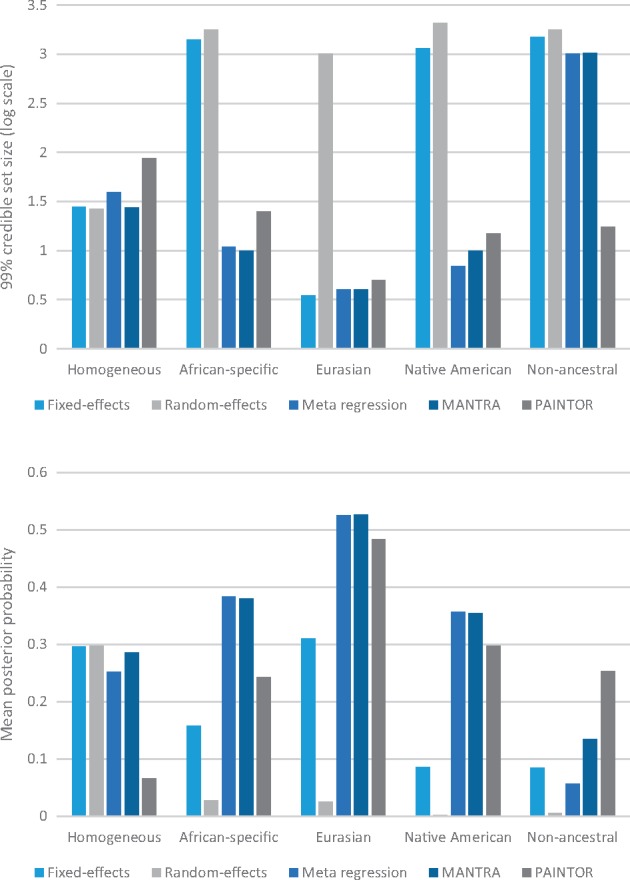
Trans-ethnic meta-analysis of genome-wide association studies (GWAS) across diverse populations can increase power to detect complex trait loci when the underlying causal variants are shared between ancestry groups. However, heterogeneity in allelic effects between GWAS at these loci can occur that is correlated with ancestry. Here, a novel approach is presented to detect SNP association and quantify the extent of heterogeneity in allelic effects that is correlated with ancestry. We employ trans-ethnic meta-regression to model allelic effects as a function of axes of genetic variation, derived from a matrix of mean pairwise allele frequency differences between GWAS, and implemented in the MR-MEGA software. Through detailed simulations, we demonstrate increased power to detect association for MR-MEGA over fixed- and random-effects meta-analysis across a range of scenarios of heterogeneity in allelic effects between ethnic groups. We also demonstrate improved fine-mapping resolution, in loci containing a single causal variant, compared to these meta-analysis approaches and PAINTOR, and equivalent performance to MANTRA at reduced computational cost. Application of MR-MEGA to trans-ethnic GWAS of kidney function in 71,461 individuals indicates stronger signals of association than fixed-effects meta-analysis when heterogeneity in allelic effects is correlated with ancestry. Application of MR-MEGA to fine-mapping four type 2 diabetes susceptibility loci in 22,086 cases and 42,539 controls highlights: (i) strong evidence for heterogeneity in allelic effects that is correlated with ancestry only at the index SNP for the association signal at the CDKAL1 locus; and (ii) 99% credible sets with six or fewer variants for five distinct association signals.
跨种族全基因组关联研究(GWAS)在不同人群中的荟萃分析,当潜在因果变异在不同祖先群体间共享时,可增强检测复杂性状位点的能力。然而,这些位点的GWAS之间等位基因效应的异质性可能会出现,且与祖先相关。本文提出了一种新方法,用于检测单核苷酸多态性(SNP)关联,并量化与祖先相关的等位基因效应的异质性程度。我们采用跨种族元回归,将等位基因效应建模为遗传变异轴的函数,该函数源自GWAS之间平均成对等位基因频率差异矩阵,并在MR-MEGA软件中实现。通过详细的模拟,我们证明,在一系列族群间等位基因效应异质性的情景下,MR-MEGA检测关联的能力比固定效应和随机效应荟萃分析更强。我们还证明,与这些荟萃分析方法和PAINTOR相比,在包含单个因果变异的位点上,MR-MEGA具有更高的精细定位分辨率,且在降低计算成本的情况下,其性能与MANTRA相当。将MR-MEGA应用于71461名个体的肾功能跨种族GWAS表明,当等位基因效应的异质性与祖先相关时,其关联信号比固定效应荟萃分析更强。将MR-MEGA应用于22086例病例和42539例对照中对四个2型糖尿病易感位点的精细定位,突出了以下两点:(i)仅在CDKAL1位点关联信号的索引SNP处,有强有力的证据表明等位基因效应的异质性与祖先相关;(ii)五个不同关联信号的99%可信集包含六个或更少的变异。