Graduate Program in Biomedical Sciences, Prince of Songkla University, Songkhla, Thailand.
Program in Translational Medicine, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand.
Sci Rep. 2017 Sep 21;7(1):12096. doi: 10.1038/s41598-017-12317-3.
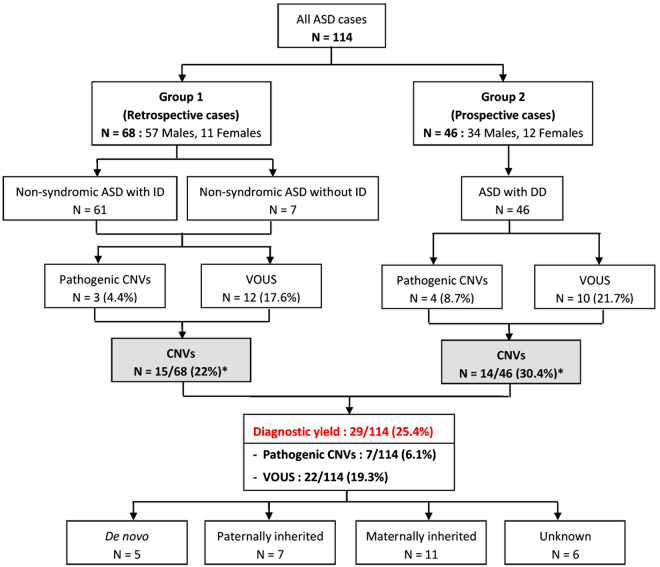
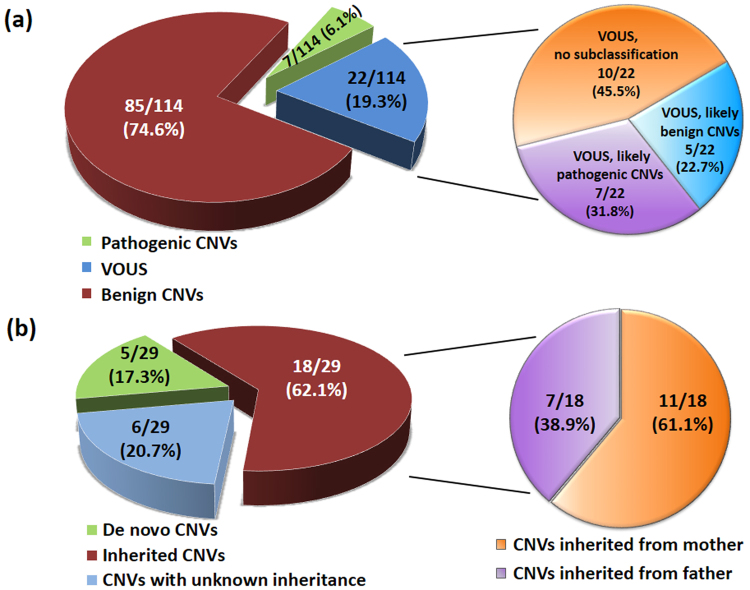
Chromosomal microarray (CMA) is now recognized as the first-tier genetic test for detection of copy number variations (CNVs) in patients with autism spectrum disorder (ASD). The aims of this study were to identify known and novel ASD associated-CNVs and to evaluate the diagnostic yield of CMA in Thai patients with ASD. The Infinium CytoSNP-850K BeadChip was used to detect CNVs in 114 Thai patients comprised of 68 retrospective ASD patients (group 1) with the use of CMA as a second line test and 46 prospective ASD and developmental delay patients (group 2) with the use of CMA as the first-tier test. We identified 7 (6.1%) pathogenic CNVs and 22 (19.3%) variants of uncertain clinical significance (VOUS). A total of 29 patients with pathogenic CNVs and VOUS were found in 22% (15/68) and 30.4% (14/46) of the patients in groups 1 and 2, respectively. The difference in detected CNV frequencies between the 2 groups was not statistically significant (Chi square = 1.02, df = 1, P = 0.31). In addition, we propose one novel ASD candidate gene, SERINC2, which warrants further investigation. Our findings provide supportive evidence that CMA studies using population-specific reference databases in underrepresented populations are useful for identification of novel candidate genes.
染色体微阵列(CMA)现在被认为是检测自闭症谱系障碍(ASD)患者拷贝数变异(CNVs)的一线遗传检测方法。本研究旨在鉴定已知和新的与 ASD 相关的 CNVs,并评估 CMA 在泰国 ASD 患者中的诊断效果。使用 Infinium CytoSNP-850K BeadChip 检测了 114 名泰国患者的 CNVs,其中 68 名是回顾性 ASD 患者(第 1 组),使用 CMA 作为二线检测方法,46 名是前瞻性 ASD 和发育迟缓患者(第 2 组),使用 CMA 作为一线检测方法。我们鉴定出 7 个(6.1%)致病性 CNVs 和 22 个(19.3%)不确定临床意义的变异(VOUS)。在第 1 组和第 2 组的 22%(15/68)和 30.4%(14/46)的患者中分别发现了总共有 29 名患者有致病性 CNVs 和 VOUS。两组之间检测到的 CNV 频率差异没有统计学意义(卡方=1.02,自由度=1,P=0.31)。此外,我们提出了一个新的 ASD 候选基因 SERINC2,值得进一步研究。我们的研究结果提供了支持性证据,表明在代表性不足的人群中使用基于人群的参考数据库进行 CMA 研究有助于鉴定新的候选基因。